
 Artículo de Acceso Abierto
Artículo de Acceso Abierto
 Este Artículo de Acceso Abierto está licenciado bajo una Licencia Creative Commons Atribución-No Comercial 3.0 Unported
Este Artículo de Acceso Abierto está licenciado bajo una Licencia Creative Commons Atribución-No Comercial 3.0 Unported
Intek Song a, Jinyoung Koob y Seok Min Yoon*c
a, Jinyoung Koob y Seok Min Yoon*c
Departamento de Química Aplicada, Universidad Nacional de Andong, 1375 Gyeongdong-ro, Andong, Gyeongbuk 36729, República de Corea
Departamento de Química, Universidad de Ciencia y Tecnología de Pohang, 77 Cheongam-ro, Pohang, Gyeongbuk 37673, República de Corea
Departamento de Bio-Nanoquímica, Universidad de Wonkwang, 460 Iksandae-ro, Iksan, Jeonbuk 54538, República de Corea. Correo electrónico: [email protected]
Publicado por primera vez el 6 de agosto de 2019
Un enlace cuádruple formado entre átomos de bloque d o bloque f es un tema de investigación interesante debido a su naturaleza única, que incluye una distancia de enlace súper corta y un estrecho espacio de energía entre δ y δ*. Entre varios complejos de enlace múltiple, los acetatos Cr(II) de enlace cuádruple se consideran útiles para controlar la brecha de energía δ–δ* por la basicidad de Lewis de ligandos adicionales. Sin embargo, la síntesis y preparación de los cristales de gran tamaño de alta calidad de acetatos Cr(II) coordinados con ligandos axiales (Cr2(OAc)4L2) han sido difíciles debido a su vulnerabilidad al O2, un agente oxidante representativo en condiciones aeróbicas. En este estudio, reportamos una síntesis fácil de cristales a escala submilimétrica de Cr2(OAc)4L2 por disolución simple de Cr2(OAc)4 en solventes de ligando L. Para obtener Cr2(OAc)4L2 ligado de forma estable, se disolvieron acetatos anhidros de Cr(II) (Cr2(OAc)4) en los solventes de ligando L, que se desgasificaron de O2 disuelto. Además, los monocristales a escala submilimétrica de Cr2 (OAc)4L2 se produjeron rápidamente durante menos de una hora mediante el proceso de secado por gota. La fase monocristalina de los complejos Cr(II) sintetizados se midió mediante técnicas de difracción de rayos X, confirmando la dependencia de la basicidad de Lewis de los ligandos axiales adicionales en la distancia de enlace cuádruple Cr-Cr. Además, se observó que los picos Raman de los enlaces cuádruples en Cr2 (OAc)4L2 se desplazaron al rojo con el aumento de la basicidad de los ligandos axiales.
Introducción
Ha habido bastante progreso en el campo de la investigación sobre enlaces múltiples entre metales de transición desde que el grupo de Algodón sintetizó el acetato de Cr(II) unido cuádruple (Cr2(OAc)4) a principios de la década de 19701,2.Más recientemente, el grupo de Potencia informó de enlaces quíntuples Cr2 además del enlace cuádruple, 3 estimulando la investigación en profundidad de los enlaces de metal a metal (M-M).4-6 El enlace M–M múltiple, particularmente el enlace cuádruple, existe porque los orbitales d o f de los metales dan lugar a un enlace delta (δ), así como a un enlace sigma (σ) y dos enlaces pi (π) (Fig. 1a y b).1,2 Dado que la formación de enlaces cuádruples requiere orbitales d, proporcionan enlaces ultracortos (∼2 Å) de metal a metal1,2 y una brecha de energía estrecha.7 Además, varios ligandos pueden coordinarse a la posición axial de un complejo enlazado múltiple M–M. Por lo tanto, la coordinación axial con el enlace M–M ha sido un tema de investigación importante para controlar la longitud del enlace M–M y su reactividad dependiendo de los tipos de ligando.3 Por lo tanto, se han utilizado enlaces cuádruples para construir una cadena unidimensional de átomos metálicos de transición, para permitir la adsorción selectiva y reversible de O2, y un catalizador eficiente prometedor.8,9 Además, Cotton et al.7 informó que su magnetismo puede ser cambiado a temperatura ambiente por la transición δ-a-δ * entre el estado singlete diamagnético (σ2π2δ2) y el estado triplete paramagnético (σ2π2δδ*).
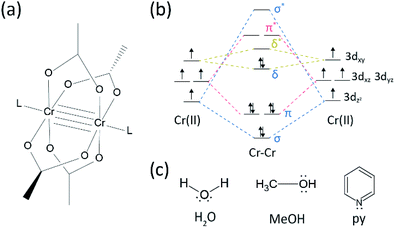
Hay varios complejos organometálicos tener delta bonos, tales como Re2Cl82−,10 Mo2(mhp)4,11 Cr2(OAc)4,12 etc.1-6 Entre ellos, ejemplos significativos son el acetato anhidro Cr(II) estructurado con rueda de paletas (Cr2(OAc)4), que carece de ligandos axiales, y sus derivados que tienen ligandos axiales (Cr2(OAC)4L2, L = ligando) (Fig. 1a).1,13 Dado que Cr (II) tiene configuración electrónica d4, dos iones Cr(II) en los acetatos llenan completamente los cuatro orbitales de enlace de un enlace cuádruple (orden de enlace = 4) (Fig. 1b). Por lo tanto, los acetatos de Cr(II) son moderadamente estables al aire a diferencia de otros compuestos de Cr(II) como el CrCl2; como dicta el diagrama de Pourbaix, 14 la mayoría de los compuestos Cr(II) son muy higroscópicos y se oxidan fácilmente a Cr(III). Además, los acetatos Cr (II) son buenos ácidos de Lewis que aceptan pares de electrones en sus posiciones axiales. Estos donantes de electrones contribuyen a los orbitales antibonding δ* vacíos (LUMO), por lo que la basicidad de Lewis de los ligandos afecta las propiedades fisicoquímicas modulando el estrecho espacio de energía entre los orbitales δ y δ*.1,12,13,15 Por lo tanto, la síntesis de acetatos Cr(II) con varios ligandos axiales L (Cr2(OAc)4L2) facilitaría el estudio fundamental de la naturaleza de los enlaces delta únicos.
Tradicionalmente, los Cr2 (OAc) 4L2 se han preparado por intercambio de ligandos del hidrato; por ejemplo, la estratificación de piridina (py) sobre una solución acuosa del hidrato (L = H2O)precipita pequeños cristales de Cr2(OAc)4(py)2.16 Sin embargo, dicha difusión interfacial puede producir cristales de Cr2(OAc)4(H2O) 2 como subproductos (Fig. S1†). Una forma bastante sencilla de coordinar los ligandos deseados es disolver Cr2 (OAc)4 en el disolvente del ligando objetivo, como señaló Cotton et al.1 Sin embargo, este método se ha restringido a pocos tipos de ligandos, como el CH3OH.7
Los acetatos Cr (II) deben manejarse con cuidado para evitar la oxidación de Cr(II) a Cr(III), ya que incluso los enlaces cuádruples no son lo suficientemente fuertes como para evitar por completo la oxidación;17 en su lugar, aceleran el proceso de oxidación en cierta medida. Esto se debe a que el O2 en el aire es un agente oxidante tan bueno que rompe efectivamente el enlace cuádruple. Por lo tanto, el oxígeno disuelto en el disolvente debe eliminarse, o desgasificarse, antes de disolver el acetato de Cr(II). Sin embargo, hasta donde sabemos, el proceso de desgasificación no se ha mencionado ni informado explícitamente.1,7,12,16
En este documento, revelamos un proceso simple de desgasificación por solvente para las diversas ligaduras del acetato anhidro Cr(II) en la posición axial, para preparar Cr2(OAc)cristalino 4L2 (L = H2O, CH3OH (MeOH) y piridina (py)) (Fig. 1c). Luego, los cristales objetivo se obtienen bajando la temperatura de la solución o el proceso de secado por goteo (Fig. 3a). Todo el proceso tarda menos de un día en producir cristales de escala submilimétrica, mientras que los métodos sintéticos convencionales tardaron varios días según las literaturas anteriores.15,18 Nuestros hallazgos contribuirían a promover la exploración de enlaces cuádruples entre átomos metálicos, la extensión de redes basadas en enlaces múltiples de metal a metal (M-M) para formar polimeros de coordinación19,20 o estructuras metal-orgánicas8,21, así como el estudio de sus propiedades físicas y químicas únicas de los enlaces cuádruples.
Resultados y discusión
Síntesis de Cr2 anhidro (ACo)4
Cr2 anhidro(ACo)4, el precursor del Cr2(ACo) 4L2 se ha preparado recociendo el hidrato o etanolato (L = EtOH) en vacío. Sin embargo, este proceso produce polvo amorfo, que se oxida fácilmente.12,17 Un enfoque alterado para preparar Cr2 (OAc)4 por una reacción química directa entre el ácido acético y el cromoceno [Cr (C2H5)2], pero el cromoceno es un complejo inflamable y sensible al aire.18,22 Por lo tanto, ambos métodos no son adecuados para preparar Cr2(OAc)4 que sea estable en el aire.
Recientemente, Levy et al.22 informó de otro método sintético para preparar el Cr2 cristalino (OAc)4, utilizando precursores estables al aire como polvo Cr (0), ácido acético, anhídrido acético y HBr. Además, el producto es estable en el aire hasta varias horas, a diferencia de los predecesores. Por lo tanto, adaptamos Levy et al.forma de preparación de los cristales de alta pureza de Cr2 (OAc)4 con ligeras modificaciones(consulte la sección Experimental para más detalles en ESI†).
En nuestro proceso sintético, se utilizó HCl concentrado (37%) (aq) en lugar de HBr o KBr, y el polvo de Cr se redujo a la mitad en la cantidad de referencia. El tiempo total de reacción fue de menos de una hora para evitar la oxidación a Cr(III). Dichas modificaciones tienen por objeto garantizar la exclusión de subproductos o reactivos no reaccionados [por ejemplo, acetato de Cr (III) y polvo de Cr(0), respectivamente] en el producto final. Por lo tanto, el polvo cristalino rojo se preparó después de simplemente filtrar el producto y lavarlo con exceso de acetona para eliminar los acetatos Cr(III) (Fig. 2a).
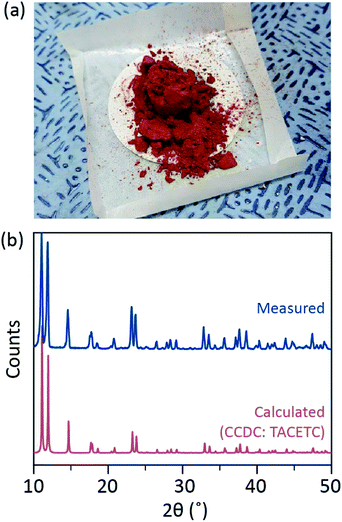
El polvo se secó al vacío a temperatura ambiente durante unas horas. Como se muestra en la Fig. 2b, polvo de difracción de rayos X (PXRD) reveló que el polvo rojo es Cr2(OAc)4 tener un triclínico de la celda unidad (P-1, a = 7.583 Å, b = 8.688 Å, c = 5.178 Å, α = 111.16°, β = 95.77°, γ = 98.16°, V = 310.626 Å3); el patrón es idéntico a un patrón calculado a partir informado anteriormente de información cristalográfica de Cr2(OAc)4 (CCDC#: TACETC).
Tenga en cuenta que el polvo obtenido se utilizó sin purificación adicional, ya que era muy difícil de purificar. Cuando se intentó la recristalización en fase de vapor de Cr2(OAc)4 para la purificación, nada se evaporó por debajo de 270 °C bajo flujo Ar. Solo una pequeña porción del polvo rojo se evaporó a temperaturas superiores a 270 ° C y inferiores a 300 ° C, y el resto se descompuso en polvo negro (Fig. S2†). El polvo evaporado se recristalizó en una película delgada rojiza, pero se descompuso tan rápidamente en verde, lo que implica oxidación a Cr(III), que no se pudo recuperar para reacciones posteriores. A una temperatura superior a 300 ° C, el polvo se descompuso completamente sin evaporación. Tal dificultad en la recristalización en fase de vapor es consistente con informes anteriores.1,13,15
Síntesis de Cr2 (OAc) 4L2
Para sintetizar los acetatos Cr(II)coordinados con ligandos axiales (Cr2(OAC) 4L2), primero seleccionamos disolventes de ligandos objetivo de la siguiente manera: MeOH, H2O y py (Fig. 1c). Estos ligandos tienen pares solitarios de electrones para formar un enlace de coordinación estable a Cr2 (OAc)4. Tienen diferente basicidad de Lewis (MeOH < H2O < py), y en consecuencia, se sabe que la distancia internuclear del enlace cuádruple en el complejo ligado se extiende desde 2.288 Å (anhidro), 2.329 Å (MeOH), 2,362 Å (H2O), y hasta 2,369 Å (py), según la basicidad de Lewis de los ligandos axiales.7,12,16,23
Los disolventes de ligando se desgasificaron calentándolos justo por debajo de su punto de ebullición con una inyección de gas Ar (Fig. 3a). Este proceso de desgasificación es un requisito previo para la disolución de Cr2 (OAc)4 en el disolvente objetivo para evitar la degradación de los enlaces cuádruples, ya que obstruye la oxidación vigorosa por el O2 disuelto. Cuando Cr2(OAc)4 se disolvió en un disolvente desgasificado, el color de la solución cambió instantáneamente a rojo como Cr2(OAc)4 (Fig. 3b). Tenga en cuenta que la disolución en H2O desgasificado no es instantánea; cuando se puso una pequeña cantidad de Cr2(OAc)4 en H2O desgasificado, al principio era azul, y luego agregar una cantidad adicional de soluto finalmente lo tornó de color marrón rojizo. Esta disolución en dos etapas puede atribuirse al equilibrio entre Cr2(ACo)4 (aq) y Cr2+ (aq), de la siguiente manera:1,24 (i) Cr2(ACo)4 2 2Cr(ACo)2, (ii) Cr(ACo)2 Cr Cr(ACo)+ + ACo−, (iii) Cr(ACo)+ Cr Cr2+ + ACo−.
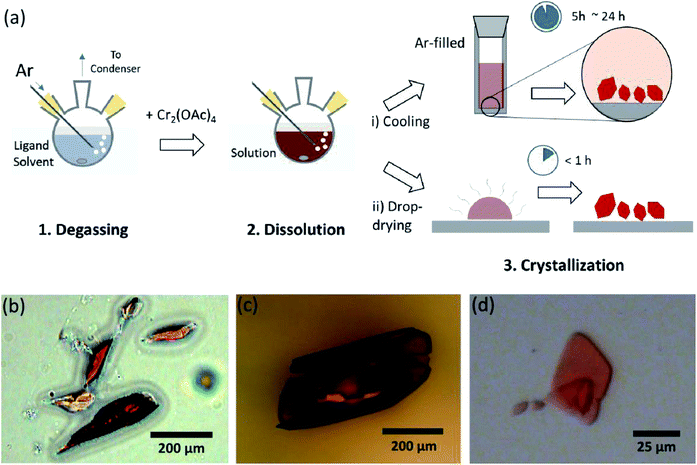
Por el contrario, cuando Cr2(OAc)4 se disolvió en disolventes no desgasificados, la solución fue azul para H2O y MeOH y naranja para piridina, que se atribuyen a la descomposición de Cr2(OAc)4 (Fig. S3†).1 Tenga en cuenta que también hay ciertos disolventes que no disuelven Cr2(OAc)4 incluso después del proceso de desgasificación. Cuando se colocó Cr2(OAc)4 en hexano o tolueno, que no tiene pares solitarios de electrones para donar, no se observó disolución ni coordinación; todo el polvo se hundió en el fondo del recipiente de reacción (Fig. S4†). Tal solubilidad selectiva muestra que la coordinación de los ligandos acompaña la disolución de Cr2 (OAc)4, y viceversa.1
Cristalización de Cr2(OAc)4L2
Cuando se terminó la ligadura en condición anóxica, se detuvo la agitación para la precipitación de reactivos restantes como polvo Cr o polvo Cr2(OAc)4 sin disolver. La temperatura se mantuvo idéntica al proceso de desgasificación y disolución (es decir, justo por debajo del punto de ebullición del disolvente del ligando). Para obtener los cristales de los complejos ligados (Cr2(OAc)4L2), el sobrenadante transparente se colocó cuidadosamente en un vial lleno de Ar para enfriarse en condiciones anóxicas a temperatura ambiente, ya que la solución es muy sensible al aire; solo una breve exposición cambia el color de rojo a azul o verde, lo que implica la oxidación del complejo. Pequeños cristales rojos en el vial lleno de Ar se habían cultivado en el fondo del vial en menos de 24 horas (proceso de cristalización (i) en la Fig. 3a). Los cristales también se pueden obtener muy rápidamente mediante el secado uniforme por gota del sobrenadante en un sustrato, y se tarda menos de una hora en condiciones ambientales (proceso de cristalización (ii) en la Fig. 3a). Los productos cristalinos por ambos métodos son idénticos; son cristales rojizos de escala submilimétrica, cuyos colores son sutilmente diferentes dependiendo de los ligandos (Fig. 3b–d).
Caracterización de Cr2 (OAC)4L2
Además, los estudios de difracción de rayos X monocristalinos(SCXRD) confirmaron que la estructura cristalográfica de los cristales Cr2 (OAc)4L2 obtenidos coincide con la de los cristales notificados anteriormente (Tabla 1).7,12,16 Tenga en cuenta que el SCXRD se intentó para la confirmación estructural del cristal único de gran tamaño en lugar del PXRD, ya que el rendimiento de los cristales no era alto (7,94% para L = H2O, 11,26% para L = MeOH y 3,70% para L = py). Tal bajo rendimiento se atribuye a que la solubilidad de Cr2(OAc)4 en los disolventes de ligantes es pobre1,y también a que la mayoría de las moléculas complejas ligadas permanecen como soluto incluso después de la cristalización. Además, a través de los estudios de SCXRD, se confirma que la distancia internuclear entre M–M es alargada dependiendo de la basicidad de Lewis de los ligandos axiales adicionales (basicidad de Lewis: MeOH < H2O < py). Por lo tanto, el orden de distancias M–M del Cr2(OAc)4L2 se divulga por el SCXRD de la siguiente manera: Cr2(OAc)4 < Cr2(OAc)4(MeOH)2 < Cr2(OAc)4(H2O)2 < Cr2(OAc)4(py)2. Además, los cristales ligados rápidamente sintetizados también fueron estables en el aire durante, pero no limitados a, 12 horas, al igual que los cristales Cr2(OAc)4 originales. Una razón probable sería el empaquetamiento de las moléculas en los cristales, lo que puede dificultar el ataque de ciertos agentes oxidantes como el O2. Estos fragmentos de evidencia nos dicen que nuestro método de cristalización simple hace que los diversos ligandos se adhieran fácilmente a la posición axial del enlace cuádruple Cr–Cr con mayor estabilidad.
| L | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | Volumen (Å3) | Nota | |
|---|---|---|---|---|---|---|---|---|---|
| H2O | 13.105 | 8.568 | 13.838 | 90.00 | 116.71 | 90.00 | 1388 | C2/c | 31 |
| MeOH | 8.065 | 7.406 | 13.296 | 90.00 | 92.84 | 90.00 | 793.2 | P21/n | 32 |
| py | 12.875 | 8.589 | 19.524 | 90.00 | 90.00 | 90.00 | 2159 | Pbca | 33 |
La espectroscopia Raman se ha utilizado como una poderosa herramienta para estudiar los modos de vibración de complejos binucleares de enlace múltiple que contienen Cr, Mo, W o Re y una correlación entre la fuerza de enlace M-M y la distancia de acuerdo con varios ligandos.1,25-27 Como se muestra en la Fig. 4, espectroscopia Raman confocal (Alpha 300R, WITec) reveló modos de vibración de los cristales de acetatos Cr(II). Los datos espectrales Raman de los acetatos Cr(II) se recogieron en menos de unos minutos, para evitar la degradación de los complejos mientras los cristales se irradian mediante una fuente de excitación láser (532 nm, 0,5 mW, Nd:YAG) en condiciones ambientales.
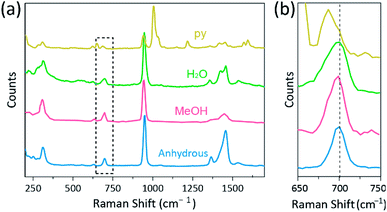
En nuestro experimento, el espectro Raman medido de Cr2(OAc)4(H2O)2 es idéntico a un informe anterior25,pero los espectros Raman de los otros complejos sintetizados, incluido el Cr2 anhidro(OAc)4, no se reportan, según nuestro mejor conocimiento. Los complejos de enlace cuádruple Cr-Cr se muestran en la Fig. 4 tienen un patrón similar del espectro Raman del Cr2 (OAc)4 (H2O) 2, posiblemente debido a la similitud de las estructuras moleculares. En los espectros Raman, se han asignado bandas de alrededor de 550 cm−1 y 350 cm−1 a los modos de estiramiento Cr–Cr y torsión/flexión de acetatos Cr(II), respectivamente.25,28,29 Sin embargo, las posiciones exactas de la banda apenas se distinguen de los espectros Raman medidos de todos los acetatos Cr(II) debido a su baja intensidad18 o ancho amplio (Fig. S5†). Como alternativa, las envolventes de armónicos de la vibración de los enlaces cuádruples Cr–Cr se muestran obviamente alrededor de 700 cm−1 (2 × ∼350 cm−1) (Fig. 4b).29 De hecho, a medida que aumenta la basicidad del ligando axial, la banda de armónicos se desplaza al rojo en consecuencia.30 Esta pista refleja que la fuerza de enlace de los enlaces cuádruples es más débil, ya que aumenta la basicidad de Lewis de los ligandos axiales.29 Además, el grado de desplazamiento al rojo de los acetatos Cr(II) en los espectros Raman se correlaciona con sus distancias Cr-Cr. Por lo tanto, la banda de armónicos de Cr2(OAc)4 muestra el número de onda más alto, pero Cr2(OAc)4(py)2 tiene el número de onda más bajo entre los complejos de enlace cuádruple Cr–Cr.
Conclusión
Desarrollamos una forma factible y rápida de sintetizar monocristales a escala submilimétrica de Cr2 (OAc)4L2, donde L = H2O, MeOH y py. La clave del éxito es la eliminación de los agentes oxidantes disueltos en los disolventes de ligando antes de disolver el polvo cristalino de Cr2 (OAc)4. Además, la disolución de Cr2 (OAc)4 en ciertos solventes de ligando ocurre solo cuando se forman enlaces de coordinación entre el acetato anhidro de Cr(II) (ácido de Lewis) y las moléculas de ligando (base de Lewis). Nuestro resultado demuestra que varias moléculas pueden coordinarse como ligandos axiales para multiplicar complejos organometálicos unidos por un proceso fácil. Dicha versatilidad y simplicidad se pueden ampliar aún más para reemplazar los ligandos de la rueda de paletas y los átomos metálicos, logrando una combinación aún más diversa para desarrollar la gran extensión del sistema de red basado en M-M, como los polímeros de coordinación en cadena M–M y los marcos metal–orgánicos. El diseño y la preparación de tales complejos novedosos contribuirían a la comprensión fundamental de los orbitales delta y los enlaces múltiples y sus aplicaciones en diversos campos de la física, la química y la ingeniería relacionada.
Conflictos de intereses
No hay conflictos que declarar.
Agradecimientos
Los autores agradecen al Prof. Dr. Minjoong Yoon de la Universidad Nacional de Chungnam y al Prof. Dr. Hee Cheul Choi de la Universidad de Ciencia y Tecnología de Pohang por sus valiosos comentarios. Este trabajo fue apoyado por la Universidad de Wonkwang en 2018.
Notas y referencias
- F. A. Cotton, C. A. Murillo y R. A. Walton, Multiple Bonds Between Metal Atoms, Springer-Verlag, Nueva York, 2005 Search PubMed.
- F. A. Cotton, B. G. DeBoer, M. D. LaPrade, J. R. Pipal and D. A. Ucko, J. Am. Chem. Soc., 1970, 92, 2926-2927 CrossRef CAS.
- T. Nguyen, A. D. Sutton, M. Brynda, J.C. Fettinger, G. J. Long y P. P. Power, Science, 2005, 310, 844-847 CrossRef CAS PubMed.
- F. R. Wagner, A. Noor y R. Kempe, Nat. Chem., 2009, 1, 529-536 CrossRef CAS PubMed.
- K. A. Kreisel, G. P. A. Yap, O. Dmitrenko, C. R. Landis y K. H. Theopold, J. Ser. Chem. Soc., 2007, 129, 14162-14163 CrossRef CAS PubMed.
- A. Falceto, K. H. Theopold and S. Alvarez, Inorg. Chem., 2015, 54, 10966-10977 CrossRef CAS PubMed.
- F. A. Cotton, H. Chen, L. M. Daniels and X. Feng, J. Am. Chem. Soc., 1992, 114, 8980-8983 CrossRef CAS.
- E. D. Bloch, W. L. Queen, M. R. Hudson, J. A. Mason, D. J. Xiao, L. J. Murray, R. Flacau, C. M. Brown and J. R. Long, Angew. Chem., Int. Eréctil., 2016, 55, 8605-8609 CrossRef CAS PubMed.
- L. J. Murray, M. Dinca, J. Yano, S. Chavan, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7856-7857 CrossRef CAS PubMed.
- F. A. Cotton and C. B. Harris, Inorg. Chem., 1965, 4, 330-333 CrossRef CAS.
- W. Clegg, C. D. Garner, L. Akhter and M. H. Al-Samman, Inorg. Chem., 1983, 22, 2466-2468 CrossRef CAS.
- Algodón A. F., Arroz C. E. y Arroz G. W., J. Am. Chem. Soc., 1977, 99, 4704-4707 CrossRef CAS.
- S. N. Ketkar and M. Fink, J. Am. Chem. Soc., 1985, 107, 338-340 CrossRef CAS.
- M. Pourbaix, Atlas de equilibrios electroquímicos en soluciones acuosas, Asociación Nacional de Ingenieros de Corrosión, 1974 Search PubMed.
- F. A. Cotton, E. A. Hillard, C. A. Murillo and H. C. Zhou, J. Am. Chem. Soc., 2000, 122, 416-417 CrossRef CAS.
- F. A. Cotton and T. R. Felthouse, Inorg. Chem., 1980, 19, 328-331 CrossRef CAS.
- J.C. Reeve, J. Chem. Educ., 1985, 62, 444 CrossRef CAS.
- Algodón A. F., M. W. Extine y Arroz G. W., Inorg. Chem., 1978, 17, 176-186 CrossRef CAS.
- H.-C. Chang, J.-T. Li, C.-C. Wang, T.-W. Lin, H.-C. Lee, G.-H. Lee y S.-M. Peng, Eur. J. Inorg. Chem., 1999, 1999, 1243-1251 CrossRef.
- M. Yang, T.-W. Lin, C.-C. Chou, H.-C. Lee, H.-C. Chang, G.-H. Lee, M. Leung and S.-M. Peng, Chem. Commun., 1997, 2279-2280 RSC.
- L. J. Murray, M. Dinca, J. Yano, S. Chavan, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7856-7857 CrossRef CAS PubMed.
- O. Levy, B. Bogoslavsky y A. Bino, Inorg. Chim. Acta, 2012, 391, 179-181 CrossRef CAS.
- F. A. Cotton, B. G. DeBoer, M. D. LaPrade, J. R. Pipal and D. A. Ucko, Acta Crystallogr., Secta. B: Estructura. Cristalogr. Cryst. Chem., 1971, 27, 1664-1671 CrossRef CAS.
- R. D. Cannon and J. S. Stillman, Inorg. Chem., 1975, 14, 2207-2214 CrossRef CAS.
- Y. M. Huang, H. R. Tsai, S. H. Lai, S. J. Lee, I. C. Chen, C. L. Huang, S. M. Peng and W. Z. Wang, J. Phys. Chem. C, 2011, 115, 13919-13926 CrossRef CAS.
- R. E. Da Re, J. L. Eglin, C. N. Carlson, K. D. John, D. E. Morris, W. H. Woodruff, J. A. Bailey, E. Batista, R. L. Martin, F. A. Cotton, E. A. Hillard, C. A. Murillo, A. P. Sattelberger and R. J. Donohoe, J. Am. Chem. Soc., 2010, 132, 1839-1847 CrossRef CAS PubMed.
- W. C. Trogler y H. B. Gray, Acc. Chem. Res., 1978, 11, 232-239 CrossRef CAS.
- F. A. Cotton, P. E. Fanwick, R. H. Niswander and J.C. Sekutowski, J. Am. Chem. Soc., 1978, 100, 4725-4732 CrossRef CAS.
- R. E. Da Re, J. L. Eglin, C. N. Carlson, K. D. John, D. E. Morris, W. H. Woodruff, J. A. Bailey, E. Batista, R. L. Martin, F. A. Cotton, E. A. Hillard, C. A. Murillo, A. P. Sattelberger and R. J. Donohoe, J. Am. Chem. Soc., 2010, 132, 1839-1847 CrossRef CAS PubMed.
- Tenga en cuenta que en el caso de Cr2(OAc)4(py)2, se asignó un pico débil cerca de 680 cm−1 como modo de armónicos en lugar del pico fuerte cerca de 650 cm−1, porque se desvía demasiado de 700 cm−1. Sin embargo, esta asignación es aceptable, porque los picos Raman de Cr2(OAc)(py)2 que son comunes a otros complejos son sustancialmente más débiles que los picos poco comunes..
- Cr2(OAc)4(H2O)2: C8H12O10Cr2, Mr = 372.18, dimensiones de cristal 0.10 × 0.09 × 0.02 mm3, monoclínico, C2/c, a = 13.105(3) Å, b = 8.5680(17) Å, c = 13.838(3) Å, β = 116.71(3)°, V = 1388.0(6) Å3, T = -173 °C, Z = 4, rcal= 1.781 g cm−3, m = 0,15 cm−1, de 1968 único reflexiones de 2076 con I > 2s(I), 94 parámetros, 2.925° < q < 29.848°, R1 = 0.0425, wR2 = 0.1183, GOF = 0.927. Depósito CCDC número 1131602..
- Cr2(OAc)4(MeOH)2: C10H18O10Cr2, Mr = 402.24, dimensiones de cristal 0.05 × 0.04 × 0.05 mm3, monoclínico, P21/n, a = 8.0650(16) Å, b = 7.4060(15) Å, c = 13.296(3) Å, β = 92.84(3)°, V = 793.2(3) Å3, T = -173 °C, Z = 2, rcal = 1.684 g cm−3, m = 0.135 cm−1, 2197 reflexiones únicas de 2277 con I > 2s(I), 105 parámetros, 3.102° < q < 29.841°, R1 = 0.0439, wR2 = 0.1198, GOF = 0.862. Refer to ref. 7..
- Cr2(OAc)4(py)2: C18H22O8N2Cr2, Mr = 496.36, crystal dimensions 0.09 × 0.08 × 0.09 mm3, orthorhombic, Pbca, a = 12.875(3) Å, b = 8.5890(17) Å, c = 19.524(4) Å, V = 2159.0(7) Å3, T = −173 °C, Z = 4, rcal= 1.533 g cm−3, m = 0.10 cm−1, 2797 unique reflections out of 3243 with I > 2s(I), 138 parameters, 2.579° < q < 29.860°, R1 = 0.0452, wR2 = 0.1206, GOF = 1.043. CCDC deposit number 1100915..
Nota de pie de página
† Información complementaria electrónica (ESI) disponible. Véase DOI: 10.1039 / c9ra04189c