
 De Acesso aberto o Artigo
De Acesso aberto o Artigo Este Acesso Aberto o Artigo está licenciado sob uma licença Creative Commons Atribuição-uso Não Comercial 3.0 Unported Licença
Este Acesso Aberto o Artigo está licenciado sob uma licença Creative Commons Atribuição-uso Não Comercial 3.0 Unported Licença
Intek Música a, Jinyoung Koob e Seok Min Yoon*c
a, Jinyoung Koob e Seok Min Yoon*c
aDepartment de Química Aplicada, Andong Universidade Nacional, 1375 Gyeongdong-ro, Andong, Gyeongbuk 36729, República da Coreia
bDepartment de Química, Pohang Universidade de Ciência e Tecnologia, 77 Cheongam-ro, Pohang, Gyeongbuk 37673, República da Coreia
cDepartment de Bio-Nano Química, Wonkwang Universidade, 460 Iksandae-ro, Iksan, Jeonbuk 54538, República da Coreia. E-mail: [email protected]
publicado pela Primeira vez em 6 de agosto de 2019
Um quádruplo vínculo formado entre d-bloco f bloco átomos é um interessante tema de pesquisa, devido à sua natureza única, incluindo um supershort colagem de distância e estreito energia de gap entre δ e δ*. Entre vários complexos ligados multiplicados, acetatos Cr(II) quadrúplamente ligados são considerados úteis para controlar o gap de Energia δ–δ* pela basicidade de Lewis de ligandos adicionais. No entanto, a síntese e preparação dos cristais de grande qualidade de acetatos Cr(II) coordenados com ligantes axiais (Cr2(OAc)4L2) têm sido difíceis devido à sua vulnerabilidade ao O2, um agente oxidante representativo em condições aeróbicas. Neste estudo, relatamos um fácil a síntese de sub-milímetro-escala de cristais de Cr2(OAc)4L2 por simples dissolução da Cr2(OAc)4 em ligante solventes L. Para obter de forma estável ligated Cr2(OAc)4L2, anidro Cr(II) acetatos (Cr2(OAc)4) foram dissolvidos em ligante solventes L, que foi desgaseificado de O2 dissolvido. Além disso, cristais simples de escala sub-milímetro de Cr2(OAc)4L2 foram produzidos rapidamente por menos de uma hora pelo processo de secagem. A fase cristalina única dos complexos CR(II) sintetizados foi medida por técnicas de difração de raios X, confirmando a dependência da basicidade de Lewis dos ligantes axiais adicionais na distância de ligação quadrúpla Cr-Cr. Além disso, observou-se que os picos Raman das ligações quádruplas em Cr2(OAc)4L2 foram Red-shifted com o aumento da basicidade dos ligantes axiais.
Introdução
tem havido algum progresso no campo da investigação em vários laços entre os metais de transição desde o Algodão grupo sintetizou o quádruplo ligada Cr(II) acetato (Cr2(OAc)4) no início da década de 1970.1,2, Mais recentemente, o Poder de grupo relatou Cr2 quíntuplo obrigações além do quádruplo de obrigações,de 3 de estimular a pesquisa em profundidade do metal-metal (M–M) de títulos.4-6 a ligação M–M múltipla, particularmente a ligação quádrupla existe porque os metais ‘ D-ou F-orbitais dão origem a uma ligação delta (δ), bem como uma ligação sigma (σ) e duas ligações pi (π) (Fig. 1a E b).1,2 uma vez que a formação de ligações quádruplas requer d-orbitais, eles fornecem ultrasort (∼2 Å) metal-metal bonds1,2 e uma estreita lacuna de energia.7 também, vários ligandos podem coordenar-se para a posição axial de um complexo de ligações múltiplas M–M. Assim, a coordenação axial para a ligação M–M tem sido um tópico de pesquisa importante para controlar o comprimento da ligação M–M e sua reatividade dependendo dos tipos de ligando.3 Portanto, ligações quádruplas têm sido usadas para construir uma cadeia unidimensional de átomos de metal de transição, para permitir adsorção seletiva e reversível de O2, e um catalisador eficiente promissor.8,9 além disso, Cotton et al.7 relataram que o magnetismo pode ser alterado em temperatura ambiente por δ para δ* a transição entre o diamagnetic estado singlete (σ2π2δ2) e o paramagnético trio de estado (σ2π2δδ*).
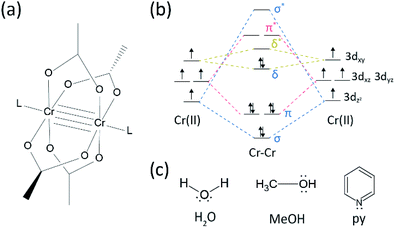
existem vários complexos organometálicos com ligações delta,tais como Re2Cl82−, 10 Mo2(mhp)4,11 Cr2(OAc)4,12 etc.Entre eles, exemplos significativos são o acetato Cr(II) anidro (Cr2(OAc)4), que não possui ligantes axiais, e seus derivados que têm ligantes axiais (Cr2(OAc)4L2, L = ligando) (Fig. 1a).1,13 desde Cr(II) tem configuração eletrônica d4, dois íons Cr (II) nos acetatos preenchem completamente os quatro orbitais de ligação de uma ligação quádrupla (ordem de ligação = 4) (Fig. 1b). Os acetatos Cr(II) são, portanto, moderadamente estáveis ao ar, ao contrário de outros compostos Cr(II), como o CrCl2.; como ditado pelo diagrama de Pourbaix, 14 compostos Cr (II) são muito higroscópicos e facilmente oxidados em Cr(III). Além disso, acetatos Cr(II) são bons ácidos de Lewis que aceitam pares de elétrons em suas posições axiais. Estes doadores de elétrons contribuem para o vazio δ * orbitais antibonding( LUMO), de modo que a base de Lewis dos ligandos afeta as propriedades físico-químicas, modulando a estreita diferença de energia entre δ e δ* orbitais.Por conseguinte, a síntese de acetatos Cr(II) com vários ligantes axiais L [Cr2 (OAc)4L2] facilitaria um estudo fundamental sobre a natureza das ligações delta únicas.
Tradicionalmente Cr2(OAc)4L2 foram preparados pelo ligante troca de hidratar; por exemplo, camadas de nucleotides (py), no topo de uma solução aquosa de hidratar (L = H2O) precipita-se pequenos cristais de Cr2(OAc)4(py)2.16 Tais interfacial difusão, no entanto, podem produzir cristais de Cr2(OAc)4(H2O)2-produtos (Fig. S1†). Uma maneira bastante reta para a frente de coordenar os ligantes desejados é dissolver Cr2(OAc)4 no solvente ligando alvo, como apontado por Cotton et al.1 no entanto, este método tem sido restrito a poucos tipos de ligandos, como CH3OH.7
os acetatos Cr(II) devem ser manuseados com cuidado para evitar a oxidação de Cr(II) A Cr(III), uma vez que mesmo as ligações quádruplas não são suficientemente fortes para evitar completamente a oxidação 17, em vez disso, acelerar o processo de oxidação em certa medida. Isto é porque O2 no ar é um agente oxidante tão bom que ele efetivamente quebra a ligação quádrupla. Por conseguinte, o oxigénio dissolvido no solvente deve ser removido ou desgaseificado antes da dissolução do acetato de Cr(II). No entanto, o processo de desgasificação não foi explicitamente mencionado nem comunicado, tanto quanto é do nosso conhecimento.1,7,12,16
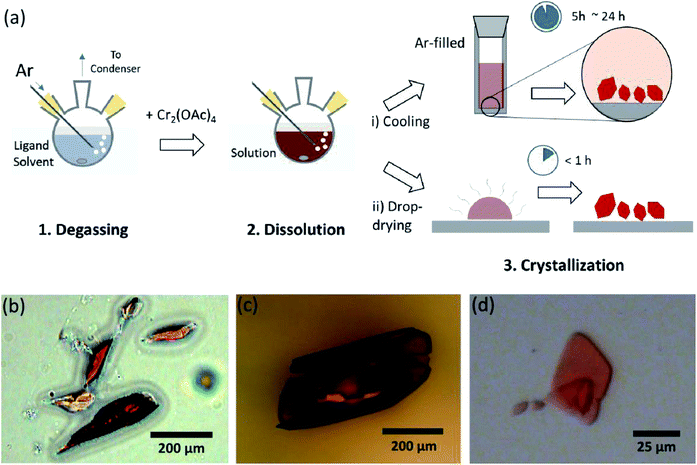
Aqui, divulgamos um simples solvente de desgaseificação do processo para os vários ligadura do anidro Cr(II) de acetato na posição axial, para preparar cristalina Cr2(OAc)4L2 (L = H2O CH3OH (MeOH), e nucleotides (py)) (Fig. 1c). Em seguida, os cristais alvo são obtidos baixando a temperatura da solução ou processo de secagem de gota (Fig. 3a). Todo o processo leva menos de um dia para produzir cristais de escala de sub-milímetros, enquanto os métodos sintéticos convencionais levaram vários dias de acordo com literaturas anteriores.15,18 Nossos resultados contribuem para promover a exploração do quádruplo ligações entre átomos de metal, a extensão do metal-metal (M–M) vários títulos baseados em redes para formar a coordenação polymers19,20 ou metal–orgânicos frameworks8,21, bem como o estudo de suas únicas propriedades físicas e químicas do quádruplo de títulos.
resultados e discussão
síntese de Cr2(OAc)anidro 4
Cr2(OAc)anidro 4, o precursor do Cr2(OAc)4L2 foi preparado por recozimento do hidrato ou etanolato (L = EtOH) no vácuo. No entanto, este processo produz um pó amorfo, que é facilmente oxidado.12,17 Um alterados abordagem à preparação de Cr2(OAc)4 por reação química entre o ácido acético e chromocene (Cr(C2H5)2), mas o chromocene ar-sensível e inflamáveis complexo.18,22 portanto, ambos os métodos não são adequados para preparar Cr2(OAc)4 que é estável no ar.
Recently, Levy et al.22 relatou outro método sintético para preparar o CR2 cristalino(OAc)4, usando precursores estáveis ao ar como o pó Cr (0), ácido acético, anidrido acético e HBr. Além disso, o produto é estável no ar até várias horas, ao contrário dos predecessores. Por conseguinte, adaptámos a Levy et al.modo de preparação dos cristais de alta pureza de Cr2 (OAc)4 com ligeira modificação (consultar a secção Experimental para mais pormenores em ESI†).
no nosso processo sintético, foi utilizado HCl (aq) concentrado (37%) em vez de HBr ou KBr, e o pó Cr foi reduzido para metade na quantidade de referência. O tempo total de reacção foi inferior a uma hora para prevenir a oxidação para Cr(III). Tais modificações destinam-se a garantir a exclusão de subprodutos ou reagentes não reagidos [por exemplo, acetato de Cr (III) e pó de Cr(0), respectivamente] no produto final. Por conseguinte, o pó cristalino vermelho foi preparado após filtrar simplesmente o produto e lavá-lo com excesso de acetona para remover acetatos Cr(III) (Fig. 2a).
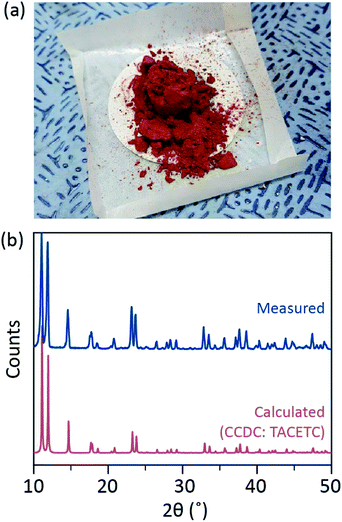
o pó foi então seco no vácuo à temperatura ambiente por algumas horas. Como mostrado na Fig. 2b, pó de difração de raios X (PXRD) revelou que o vermelho em pó é Cr2(OAc)4 ter um triclinic célula unitária (P-1, a = 7.583 Å, b = 8.688 Å, c = 5.178 Å, α = 111.16°, β = 95.77°, γ = 98.16°, V = 310.626 Å3); o padrão é idêntico a um padrão calculado a partir do relatado anteriormente cristalográfica informações de Cr2(OAc)4 (CCDC#: TACETC).Note-se que o pó obtido foi utilizado sem purificação adicional, uma vez que era muito difícil de purificar. Quando a recristalização de fase vapor de Cr2 (OAc)4 foi tentada para a purificação, nada foi evaporado abaixo de 270 °C sob o fluxo Ar. Apenas uma pequena porção do pó vermelho evaporou a uma temperatura acima de 270 ° C e abaixo de 300 °C, e o remanescente foi decomposto em pó negro (Fig. S2†). O pó evaporado foi recristalizado em película fina avermelhada, mas foi tão rapidamente decomposto em verde-implicando oxidação A Cr(III)-que não pôde ser recuperado para novas reações. A uma temperatura superior a 300 ° C, O pó foi completamente decomposto sem evaporação. Tal dificuldade na recristalização de fase vapor é consistente com relatórios anteriores.1,13,15
síntese de Cr2 (OAc)4L2
a fim de sintetizar os acetatos Cr (II)coordenados com ligantes axiais (Cr2(OAC) 4L2), selecionamos pela primeira vez solventes de ligante alvo como segue: MeOH, H2O, e py (Fig. 1c). Estes ligandos têm pares solitários de elétrons para formar uma ligação de coordenação estável para Cr2 (OAc)4. Eles têm diferentes basicidade de Lewis (MeOH < H2O < py), e, consequentemente, a distância internuclear da ligação quádrupla no complexo ligado é conhecida por se estender a partir de 2.288 Å (anidro), 2.329 Å (MeOH), 2.362 Å (H2O), e 2.369 Å (py), de acordo com a basicidade de Lewis dos ligandos axiais.7,12,16,23
os solventes ligando foram degradados aquecendo-o logo abaixo do seu ponto de ebulição com uma injecção de gás Ar (Fig. 3a). Este processo de desgaseificação é pré-requisito para a dissolução do Cr2(OAc)4 no solvente alvo para evitar a degradação das ligações quádruplas, como obstruindo a oxidação vigorosa pelo O2 dissolvido. Quando o Cr2(OAc)4 foi dissolvido num solvente desgaseirado, a cor da solução mudou instantaneamente para vermelho como o Cr2(OAc)4 (Fig. 3b). Note que a dissolução em H2O desgaseificado não é instantânea; quando uma pequena quantidade de Cr2 (OAc)4 foi colocada em H2O desgaseificado, era azul no início, e, em seguida, adicionando uma quantidade extra do soluto eventualmente tornou-o castanho avermelhado. Essas duas etapas de dissolução pode ser atribuído ao equilíbrio entre Cr2(OAc)4 (aq) e Cr2+ (aq), como segue:1,24 (i) Cr2(OAc)4 ⇄ falhas 2cr(OAc)2, (ii) Cr(OAc)2 ⇄ Cr(OAc)+ + OAc−, (iii) e Cr(OAc)+ ⇄ Cr2+ + OAc−.
Em contraste, quando Cr2(OAc)4, foi dissolvido em não-desgaseificado solventes, a solução foi azul para H2O e MeOH e laranja para nucleotides, que são atribuídos à decomposição de Cr2(OAc)4 (Fig. S3†).1 Note que existem também certos solventes que não dissolvem Cr2(OAc)4 mesmo após o processo de desgaseificação. Quando o Cr2 (OAc)4 foi colocado em hexano ou tolueno, que não tem pares de elétrons Solitários para doar, não foi observada dissolução nem coordenação.; todo o pó afundou – se até ao fundo do recipiente de reacção (Fig. S4†). Tal solubilidade seletiva mostra que a coordenação dos ligantes acompanha a dissolução do Cr2(OAc)4, e vice-versa.1
cristalização de Cr2(OAc)4L2
quando a ligação em estado anóxico foi terminada, a agitação foi interrompida por precipitação de reagentes remanescentes, como pó Cr ou pó Cr2 (OAc)4 não dissolvido. A temperatura foi mantida idêntica ao processo de desgaseificação e dissolução (isto é, logo abaixo do ponto de ebulição do solvente ligante). Para obter os cristais da ligated complexos (Cr2(OAc)4L2), limpar o sobrenadante foi cuidadosamente colocado em um Ar cheio de frasco para resfriamento sob condição anóxica, em temperatura ambiente, já que a solução é muito ar e minúsculas, apenas a uma breve exposição altera a cor de vermelho para azul ou verde, o que implica a oxidação do complexo. Pequenos cristais vermelhos no frasco cheio de Ar foram cultivados no fundo do frasco menos de 24 horas [processo de cristalização (i) na Fig. 3a). Os cristais também podem ser obtidos muito rapidamente, mesmo secando o sobrenadante em um substrato, e demora menos de uma hora sob a condição ambiente (processo de cristalização (ii) na Fig. 3a). Os produtos cristalinos por ambos os métodos são idênticos; eles são de escala sub-milimétrica, cristais avermelhados, dos quais cores são sutilmente diferentes, dependendo dos ligandos (Fig. 3b-d).
caracterização de Cr2 (OAC)4L2
outros estudos de difração de raios-X de um único cristal(SCXRD) confirmaram que a estrutura cristalográfica dos cristais CR2 (OAc)4L2 obtidos é compatível com os cristais previamente notificados (Tabela 1).7,12,16 Note que SCXRD foi tentada estrutural de confirmação de grande porte de um único cristal em vez de PXRD, desde que o rendimento dos cristais não foram altas (7.94% para L = H2O, 11.26% para L = MeOH, e 3.70% para L = py). Tal baixo rendimento é atribuído a que a solubilidade de Cr2 (OAc)4 nos solventes ligando é pobre, 1 e também que a maioria das moléculas complexas ligadas permanecem como soluto mesmo após a cristalização. Além disso, através dos estudos SCXRD, confirma–se que a distância internuclear entre M-M é alongada dependendo da basicidade de Lewis dos ligandos axiais adicionais (Lewis basicity: MeOH < H2O < py). Assim, a ordem de M–M distâncias da Cr2(OAc)4L2 são divulgados pelo SCXRD da seguinte forma: Cr2(OAc)4 < Cr2(OAc)4(MeOH)2 < Cr2(OAc)4(H2O)2 < Cr2(OAc)4(py)2. Além disso, os cristais ligados rapidamente sintetizados também foram estáveis no ar por, mas não limitado por, 12 horas, assim como os cristais originais Cr2(OAc)4. Uma razão provável seria o empacotamento das moléculas nos cristais, que podem impedir o ataque por certos agentes oxidantes como O2. Estes fragmentos de evidência nos dizem que nosso método de cristalização simples faz com que os diversos ligandos prontamente se apeguem à posição axial da ligação quádrupla Cr com maior estabilidade.
| L | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | Volume (Å3) | Espaço do grupo | Nota |
|---|---|---|---|---|---|---|---|---|---|
| H2O | 13.105 | 8.568 | 13.838 | 90.00 | 116.71 | 90.00 | 1388 | C2/c | 31 |
| MeOH | 8.065 | 7.406 | 13.296 | 90.00 | 92.84 | 90.00 | 793.2 | P21/n | 32 |
| py | 12.875 | 8.589 | 19.524 | 90.00 | 90.00 | 90.00 | 2159 | Pbca | 33 |
a espectroscopia Raman tem sido usado como uma ferramenta poderosa para o estudo de modos de vibração de multiplicar-ligado binuclear complexos contendo Cr, Mo, W, ou Re e uma correlação entre a M–M força de adesão e a distância de acordo com vários ligantes.1,25-27, como indicado na Fig. 4, confocal Raman spectroscopy(Alpha 300R, WITec) revealed vibration modes of the crystals of Cr (II) acetates. Os dados espectrais Raman de acetatos Cr(II) foram coletados menos de alguns minutos, para evitar a degradação dos complexos, enquanto os cristais foram irradiados por fonte de excitação por laser (532 nm, 0,5 mW, Nd:YAG) sob a condição ambiente.
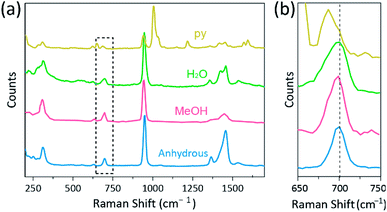
Na nossa experiência, a medida do espectro de Raman de Cr2(OAc)4(H2O)2 é idêntico ao anterior relatório,25, mas os espectros Raman dos outros sintetizados complexos, incluindo anidro Cr2(OAc)4 não são reportados, para o melhor de nosso conhecimento. Os complexos quadrúpedes Cr–Cr são apresentados na figura. 4 têm um padrão similar do espectro Raman do Cr2(OAc)4 (H2O)2, possivelmente devido à semelhança das estruturas moleculares. Nos espectros Raman, Bandas de cerca de 550 cm – 1 e 350 cm−1 foram atribuídas aos modos Cr–Cr de estiramento e torção/flexão de acetatos Cr(II), respectivamente.25,28,29 no entanto, as posições exactas da banda Mal se distinguem dos espectros de Raman medidos de todos os acetatos Cr (II) devido à sua baixa intensidade18 ou largura larga (Fig. S5†). Em alternativa, os envelopes de sobretona das ligações quadruplas Cr–Cr são obviamente exibidos em torno de 700 cm – 1 (2 × ∼350 cm−1) (Fig. 4b).29 Na verdade, à medida que a basicidade do ligando axial aumenta, a banda de sobretona é RED-deslocada em conformidade.30 Esta pista reflete que a força de ligação das ligações quádruplas sendo mais fraca, como aumentando a basicidade de Lewis dos ligandos axiais.29 Also, the degree of red-shifts of the Cr(II) acetates in Raman spectra is correlated to its Cr–Cr distances. Assim, a banda de ultrapassagem de Cr2(OAc)4 mostra o maior número de ondulações, mas Cr2(OAc)4 (py)2 tem o menor número de ondulações entre os complexos quadrúpedes Cr–Cr quadruplicamente ligados.
Conclusion
We developed a feasible and rapid way to synthesize sub-milímetro-scale single crystals of Cr2 (OAc)4L2, where L = H2O, MeOH, and py. A chave para o sucesso é a remoção de agentes oxidantes dissolvidos nos solventes ligando antes da dissolução do pó cristalino Cr2(OAc)4. Além disso, a dissolução do Cr2(OAc)4 em certos solventes ligando ocorre apenas quando as ligações de coordenação são formadas entre o acetato de cr(II) anidro (ácido de Lewis) e as moléculas ligando (base de Lewis). Nosso resultado demonstra que várias moléculas podem se coordenar como ligantes axiais para multiplicar complexos organometálicos ligados por um processo facil. Tal versatilidade e simplicidade podem ser expandidas para substituir os ligantes de roda de remo e átomos de metal, alcançando uma combinação ainda mais diversificada para desenvolver a grande extensão do sistema de rede baseado em M–M, tais como polímeros de coordenação acorrentados M–M e frameworks metal–orgânicos. O Design e a preparação de tais complexos novos contribuiriam para a compreensão fundamental dos orbitais delta e múltiplas ligações e suas aplicações em vários campos da física, química e engenharia relacionada.
conflitos de interesses
não há conflitos a declarar.
agradecimentos
os autores apreciam o Prof. Dr. Minjoong Yoon na Universidade Nacional de Chungnam e o Prof. Dr. Hee Cheul Choi na Universidade de Ciência e Tecnologia de Pohang por comentários valiosos. Este trabalho foi apoiado pela Universidade Wonkwang em 2018.
notas e referências
- F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple Bonds Between Metal Atoms, Springer-Verlag, New York, 2005 Search PubMed.Cotton, B. G. DeBoer, M. D. LaPrade, J. R. Pipal and D. A. Ucko, J. Am. Chem. Soc., 1970, 92, 2926-2927 CrossRef CAS.
- T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long and P. P. Power, Science, 2005, 310, 844-847 CrossRef CAS PubMed.
- F. R. Wagner, A. Noor and R. Kempe, Nat. Chem., 2009, 1, 529-536 CrossRef CAS PubMed.
- K. A. Kreisel, G. P. A. Yap, O. Dmitrenko, C. R. Landis e K. H. Theopold, J. Manha. Chem. Soc., 2007, 129, 14162-14163 CrossRef CAS PubMed.
- A. Falceto, K. H. Theopold and S. Alvarez, Inorg. Chem., 2015, 54, 10966-10977 CrossRef CAS PubMed.Cotton, H. Chen, L. M. Daniels and X. Feng, J. Am. Chem. Soc., 1992, 114, 8980-8983 CrossRef CAS.
- E. D. Bloch, W. L. Rainha, M. R. Hudson, J. A. Mason, D. J. Xiao, L. J. Murray, R. Flacau, C. M. Brown e J. R. Longa, Angew. Chem., T. Disfuncao., 2016, 55, 8605-8609 CrossRef CAS PubMed.
- L. J. Murray, M. Dinca, J. Yano, S. Chavan, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7856-7857 CrossRef CAS PubMed.
- F. A. Cotton and C. B. Harris, Inorg. Chem., 1965, 4, 330-333 CrossRef CAS.
- W. Clegg, C. D. Garner, L. Akhter and M. H. Al-Samman, Inorg. Chem., 1983, 22, 2466-2468 CrossRef CAS.
- F. A. Cotton, C. E. Rice and G. W. Rice, J. Am. Chem. Soc., 1977, 99, 4704-4707 CrossRef CAS.
- S. N. Ketkar e M. Fink, J. Am. Chem. Soc., 1985, 107, 338-340 CrossRef CAS.
- M. Pourbaix, Atlas of electrochemical equilibria in aqueous solutions, National Association of Corrosion Engineers, 1974 Search PubMed.Cotton, E. A. Hillard, C. A. Murillo and H. C. Zhou, J. Am. Chem. Soc., 2000, 122, 416-417 CrossRef CAS.
- F. A. Cotton and T. R. Felthouse, Inorg. Chem., 1980, 19, 328-331 CrossRef CAS.
- J. C. Reeve, J. Chem. Educ., 1985, 62, 444 CrossRef CAS.Cotton, M. W. Extine and G. W. Rice, Inorg. Chem., 1978, 17, 176-186 CrossRef CAS.
- H.-C. Chang, J.-T. Li, C.-C. Wang, T.-W. Lin, H.-C. Lee, G.-H. Lee and S.-M. Peng, Eur. J. Inorg. Chem., 1999, 1999, 1243-1251 CrossRef.* M. Yang, T.-W. Lin, C.-C. Chou, H.-C. Lee, H.-C. Chang, G.-H. Lee, M. Leung and S.-M. Peng, Chem. Comun., 1997, 2279-2280 RSC.
- L. J. Murray, M. Dinca, J. Yano, S. Chavan, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7856-7857 CrossRef CAS PubMed.
- O. Levy, B. Bogoslavsky and A. Bino, Inorg. Chim. Acta, 2012,391, 179-181 CrossRef CAS.Cotton, B. G. DeBoer, M. D. LaPrade, J. R. Pipal and D. A. Ucko, Acta Crystallogr., Seita. B: Struct. Crystallogr. Cryst. Chem., 1971,27, 1664-1671 CrossRef CAS.
- R. D. Cannon and J. S. Stillman, Inorg. Chem., 1975, 14, 2207-2214 CrossRef CAS.Huang, H. R. Tsai, S. H. Lai, S. J. Lee, I. C. Chen, C. L. Huang, S. M. Peng e W. Z. Wang, J. Phys. Chem. C, 2011, 115, 13919-13926 CrossRef CAS.
- R. E. Da Re, J. L. Eglin, C. N. Carlson, K. D. João, D. E. Morris, W. H. Woodruff, J. A. Bailey, E. Batista, R. L. Martin, F. A. de Algodão, E. A. Hillard, C. A. Murillo, A. P. Sattelberger e R. J. Donohoe, J. Am. Chem. Soc., 2010, 132, 1839-1847 CrossRef CAS PubMed.
- W. C. Trogler and H. B. Gray, Acc. Chem. Res., 1978, 11, 232-239 CrossRef CAS.Cotton, P. E. Fanwick, R. H. Niswander e J. C. Sekutowski, J. Am. Chem. Soc., 1978, 100, 4725-4732 CrossRef CAS.
- R. E. Da Re, J. L. Eglin, C. N. Carlson, K. D. João, D. E. Morris, W. H. Woodruff, J. A. Bailey, E. Batista, R. L. Martin, F. A. de Algodão, E. A. Hillard, C. A. Murillo, A. P. Sattelberger e R. J. Donohoe, J. Am. Chem. Soc., 2010, 132, 1839-1847 CrossRef CAS PubMed.
- Note que no caso de Cr2(OAc)4(py)2 um fraco pico perto de 680 cm−1 foi atribuída como o tom modo, em vez de o pico forte perto de 650 cm−1, pois muito se desvia de 700 cm−1. Esta atribuição é, no entanto, aceitável, porque os picos Raman de Cr2(OAc) (py) 2 que são comuns a outros complexos são substancialmente mais fracos do que os picos pouco comuns..
- Cr2(OAc)4(H2O)2: C8H12O10Cr2, Mr = 372.18, cristal dimensões de 0,10 × 0.09 × 0.02 mm3, monoclinic, C2/c, a = 13.105(3) Å, b = 8.5680(17) Å, c = 13.838(3) Å, b = 116.71(3)°, V = 1388.0(6) Å3, T = -173 °C, Z = 4, rcal= 1.781 g cm−3, m = 0.15 cm−1, 1968 única reflexões de 2076, com I > 2s(I), 94 parâmetros, 2,925 de° < q < 29.848°, R1 = 0.0425, wR2 = 0.1183, GOF = 0.927. Depósito CCDC número 1131602..
- Cr2(OAc)4(MeOH)2: C10H18O10Cr2, Mr = 402.24, cristal dimensões 0.05 × 0.04 × 0.05 mm3, monoclinic, P21/n, a = 8.0650(16) Å, b = 7.4060(15) Å, c = 13.296(3) Å, b = 92.84(3)°, V = 793.2(3) Å3, T = -173 °C, Z = 2, rcal = 1.684 g cm−3, m = 0.135 cm−1, 2197 única reflexões de 2277 com I > 2s(I), 105 parâmetros, 3.102° < q < 29.841°, R1 = 0.0439, wR2 = 0.1198, GOF = 0.862. Refer to ref. 7..
- Cr2(OAc)4(py)2: C18H22O8N2Cr2, Mr = 496.36, crystal dimensions 0.09 × 0.08 × 0.09 mm3, orthorhombic, Pbca, a = 12.875(3) Å, b = 8.5890(17) Å, c = 19.524(4) Å, V = 2159.0(7) Å3, T = −173 °C, Z = 4, rcal= 1.533 g cm−3, m = 0.10 cm−1, 2797 unique reflections out of 3243 with I > 2s(I), 138 parameters, 2.579° < q < 29.860°, R1 = 0.0452, wR2 = 0.1206, GOF = 1.043. CCDC deposit number 1100915..
Nota de rodapé
† informações suplementares electrónicas (ESI) disponíveis. Ver DOI: 10.1039 / c9ra04189c