Introduction
The Limb-Girdle Muscular Dystrophies (Lgmds) include a heterogenous group of disorders character by the proximal limb-girdles muscles (1, 2). Lgmd: t osoittavat Inter-ja intrafamilial variability, vaihtelevat hyvin lievistä muodoista vakaviin, varhain alkaviin, nopeasti eteneviin fenotyyppeihin (2). LGMDs voidaan luokitella autosomaalisesti dominoivaan (lgmd1) tai resessiiviseen (lgmd2). Ensimmäinen ryhmä on yleensä täysi-ikäinen, eivätkä ne ole kovin yleisiä (<10% kaikista Lgmd:istä), kun taas jälkimmäiset ovat yleisempiä (1: 15000) (1). Resessiivisistä muodoista lgmd2a (tai kalpapainopatia) on maailmanlaajuisesti yleisin LGMD-tauti, joka vaikuttaa noin 30%: iin kaikista LGMDs-tapauksista (3). Taudin kliinisiä merkkejä ovat varpaankävely, kahlaava kävely, vaikeus juoksussa ja portaiden kiipeämisessä, lapaluun taipuminen. Nivelten supistuksia, akillesjänteen lyhentymistä ja skolioosia havaitaan yleisesti, kun taas kasvojen ja kaulan lihaksia ei vaikuteta (4). Oireetonta Hyperckemiaa (5-80 kertaa normaali CPK-taso) nuorilla potilailla pidetään prekliinisenä tautivaiheena, ja se voi jatkua useita vuosia (1, 5). Lihasheikkouden alkamisikä on yleensä 15 vuotta, joskin se voi ilmaantua aikaisemmin (<12 vuotta) tai myöhemmin (>30 vuotta). Taudin eteneminen voi johtaa väijytyksen menetykseen, hengitysvajaukseen ja keuhkojen elintärkeän kapasiteetin vähenemiseen pitkälle edenneissä vaiheissa (6). Sydämeen kohdistuvia vaikutuksia (sydämen rytmihäiriöitä, sydämen johtumishäiriöitä, vasemman kammion ejektiohäiriöitä) raportoidaan vain satunnaisesti (6, 7).
kalpapainopatian diagnoosi varmistuu, kun capn3: ssa (15q15.1) (4) havaitaan patogeenisiä mutaatioita, jotka koodaavat erilaisia vaihtoehtoisesti yhdistettyjä transkriptejä. Täyspitkä transkriptio ilmaistaan kuitenkin ensisijaisesti lihaskudoksessa (8). Koodattu proteiini (CAPN3) kuuluu ei-lysosomaaliseen Ca++-riippuvaiseen kysteiiniproteaasiperheeseen. Lihaksissa CAPN3 osallistuu ”sarcomere remodelingiin”, joka on välttämätön lihasten sopeutumiselle ja kasvulle vastauksena toiminnallisiin ja metabolisiin vaatimuksiin. Tähän mennessä CAPN3: ssa on kuvattu yli 490 patogeenistä mutaatiota, joista suurin osa on yhden nukleotidin muutoksia (4). CAPN3: n mutaatiot ovat liittyneet mitokondrioiden poikkeavuuksiin, kasvuhäiriöön, lisääntyneeseen oksidatiiviseen stressiin ja sarkoomeeriseen epäjärjestykseen, jotka kaiken kaikkiaan edistävät sitä, että lihas ei pysty pitämään kuormia, aiheuttaen siten myofiberin rappeutumista ja lihasten kuihtumista (8). Kalpapainopatian diagnosointi voi olla haastavaa geneettisen heterogeenisyyden ja kliinisen ja instrumentaalisen kuvion epäspesifisyyden vuoksi. Lihasheikkous/atrofia, hypertrofia/pseudo-hypertrofia ja jänne-kontraktuurat ovat hyvin usein yhteisiä muiden LGMDs: n tai hermo-lihassairauksien kanssa. Kalpapainopatian lihasbiopsia on yleensä epäspesifinen, vaihtelevat lievistä lihaspoikkeavuuksista vakaviin dystrofisiin muutoksiin. Lisäksi immunohistokemialliset / biokemialliset markkerit eivät yleensä ole luotettavia: kalvainsignaali voi olla normaali myös toimimattoman proteiinin läsnä ollessa, ja päinvastoin se voi pienentyä muissakin kalvainopatiasta poikkeavissa lihasdystrofioissa.
siksi on suositeltavaa tehdä erotusdiagnoosi tarkkojen ja luotettavien tulosten saamiseksi. Tätä tarkoitusta varten on kehitetty molekyyligeneettisiä testausmenetelmiä, jotka vahvistavat kalpapainopatian diagnoosin, mukaan lukien multigeenipaneeli, laaja genomitutkimus (exome/genome sequencing) ja yhden geenin analyysi (direct sequencing) (9). Seuraavan sukupolven sekvensoinnin (NGS) käyttöönotto auttoi tuottamaan informatiivista tietoa, jota voidaan soveltaa diagnostisiin, ennakoiviin tai terapeuttisiin tarkoituksiin (10-12). NGS-geenipaneelit perustuvat tiettyyn sairauteen tai siihen liittyvien häiriöiden ryhmään liittyvien geenien analyysiin, jolle on ominaista geneettinen ja fenotyyppinen heterogeenisuus. NGS-paneelit voivat myös olla hyödyllisiä havaittaessa sekoittuneita tai monimutkaisia fenotyyppejä, jotka johtuvat useamman kuin yhden geenivirheen periytymisestä vanhemmilta (13). Yleensä potilaat, joilla esiintyy monimutkaisia fenotyyppejä, jotka ovat negatiivisia tunnetuille mutaatioille räätälöidyissä diagnostisissa geenipaneeleissa ja vaativat laajan differentiaalidiagnoosin, ovat oikeutettuja koko eksomeen tai genomin sekvensointiin. Koko eksome / genomin sekvensointi ovat kalliimpia lähestymistapoja tiedonhallinnan, tulkinnan ja analyyttisten kustannusten kannalta, mutta lisäävät todennäköisyyttä antaa molekyylidiagnoosi epäillylle geneettiselle häiriölle (13). Lgmd: iden osalta suositellaan erittäin paljon erityisiä NGS-paneeleita, joilla varmistetaan korkeat diagnostiset nopeudet, optimaalinen kattavuus, herkkyys ja spesifisyys kliinisessä testauksessa (9). NGS-paneelit ovat siten yksi parhaista järjestelmistä differentiaalidiagnostiikan helpottamiseksi, uusien aiheuttavien mutaatioiden tunnistamiseksi ja genotyyppi-fenotyyppi-korrelaatioiden selventämiseksi (14, 15).
tässä käsikirjoituksessa kerrotaan lgmd2a-tautia sairastavan potilaan tapauksesta, jonka osalta NGS-paneelin soveltaminen mahdollisti paitsi kalpapainopatian diagnoosin vahvistamisen (CAPN3: n mutaatiot) myös uuden mutaation tunnistamisen lmna-geenissä, joka liittyy laajentuneeseen kardiomyopatiaan. Näiden tulosten perusteella analyysi laajennettiin koskemaan probandin perheenjäseniä, jotta patologisten fenotyyppien analyysituloksia voitaisiin tulkita kattavammin.
tapauksen esittely
Probandin ja sukulaisten kliininen Luonnehdinta
probandin taudin puhkeamiseen viitattiin 10-vuotiaana, kun hän raportoi vasikan liikakasvusta, johon liittyi varpaankävelyä, vaikeuksia juoksussa, portaiden kiipeämisessä ja seisomisessa. Oireet pahenivat hitaasti, mutta etenevästi, ja myöhemmin ne liittyivät proksimaaliseen yläraajaan. Iässä 13, korkea CPK (~8000 UI/L) havaittiin muuten, ei koskaan liittynyt myoglobinuria. Potilas kävi läpi useita neuromuskulaarisia tutkimuksia eri erikoistuneissa keskuksissa. Tehtiin kaksi lihasbiopsiaa, joissa näkyi klassinen dystrofinen kuva (hypertrofiset/atrofiset kuidut, sisäiset ytimet, nekroottiset kuidut, sidekudoksen lisääntyminen), joilla ei ollut spesifisiä ominaisuuksia. Kuten odotettiin, immunokemia-ja immunoblotitutkimukset olivat tuloksettomia, mikä osoitti poikkeavan/vähentyneen α, β, γ sarkoglykaanin, β-dystroglykaanin ja dystrofiinisignaalin vain nekroottisissa kuiduissa (toistuva immunokemia johti normaaliin, myös laminiinille); dystrofiinin ja kalpaiinin immunoblotit paljastivat normaalit signaalit. Calpain – 3-immunoblotin tiedetään kuitenkin olevan ei – täysin herkkä lgmd2a-diagnoosille, sillä 20-30 prosentissa tapauksista esiintyy normaali määrä proteiinia (1, 16-18). Kliinisestä esitystavasta (hyperCKmia, vasikan hypertrofia/pseudo-hypertrofia) johtuen dystrofinopatiat suljettiin ensin pois. Lisäksi DMD: n (Xp21), FKRP: n (19q13.32) ja DYSF: n (2p13.2) geenien analyysi ei paljastanut patogeenisiä mutaatioita. Potilas tuli havaintoomme 30-vuotiaana ja esitti aksiaalista ja vyötä sekä ylä-että alaraajassa, jossa oli merkittävää kahlaavaa kävelyä ja siivekkäitä lapaluita. Alaraajoissa heikkous oli näkyvästi kvadriceps-ja glutei-lihaksissa, jotka olivat merkittävästi ipo-atrofisia, sekä todisteet” ilmeisesti hypertrofisista ” (pseudo-hypertrofiasta) pohjelihaksista (Kuva 1). Kramppeja, myalgioita ja väreilyä ei havaittu. Täydellisissä pneumologisissa (spirometria ja polysomnografia) ja kardiologisissa (kaikukuvaus, 24 h-EKG Holter-seuranta, sydämen magneettikuvaus, täydellinen Kardiologinen fyysinen tarkastus ja kohdennettu sairaushistoria sekä sydän-ja verisuonirefleksianalyysi) tutkimuksissa ei havaittu merkittäviä poikkeavuuksia. Olemme myös sulkeneet pois happaman maltaasin puutos (normaali entsyymiaktiivisuus määritykset lymfosyyttien/leukosyyttien). Lihasten magneettikuvaus (MK) osoitti, että lapaluun vyöt, nikamanväliset lihakset, reiden posterioriset lihakset (Sartorius-ja gracilis-lihakset säästyivät suhteellisesti) ja jalka (erityisesti gastrocnemius medialis ja soleus) olivat merkittävässä osassa (kuva 2). Tätä vaikutusmallia on jo raportoitu kirjallisuudessa, joka koskee magneettikuvausta kalpainopatiassa (19, 20). Lisäksi MRI-tutkimus osoitti, että pohjelihakset olivat tehokkaasti atrofisia ja niille oli ominaista rasvan tunkeutuminen, mikä aiheutti lihasten ”laajentumisen.”Kliininen esitystapa, alkamisikä, etenemisnopeus, lihasheikkouden jakautuminen ja MAGNEETTIKUVAUSLÖYDÖKSET probandissa olivat täysin yhdenmukaisia kirjallisuudessa laajasti kuvatun lgmd2a-diagnoosin kanssa (4, 21, 22).
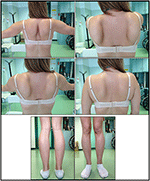
Kuva 1. Proband kliiniset ominaisuudet: siivekäs lapaluut ja yhdistelmä reiden atrofia ja vasikan pseudo-hypertrofia.
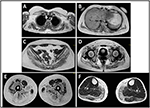
kuva 2. Proband lihas MRI: yläraajojen vyöt ja paravertebraaliset lihakset (A, B), alaraajojen vyöt ja reiden takaosa (C–E, enimmäkseen glutei) sekä Sartorius-ja gracilis-säärten takaosa (F, enimmäkseen gastrocnemius medialis/soleus).
probandin perheenjäsenten arvioinnissa ei tullut esiin mitään neuromuskulaarista sairautta eikä näyttöä vanhempien välisestä verisukulaisuudesta. Probandin äiti esitti kuitenkin 55-vuotiaana 2 synkopaalijaksoa, joiden vuoksi hänellä diagnosoitiin idiopaattinen bradykardia (jopa 40 bpm yöllä). Laajat kardiologiset tutkimukset proband diagnosoi oireisen eteis-kammiokatkoksen (AV) useiden synkopaalijaksojen/lipothymian esiintymisen jälkeen. Analyysi osoitti, että potilaalla oli ensimmäisen asteen AV-katkos, johon liittyi useita vaikean bradykardian jaksoja, jotka olivat enimmäkseen yöllisiä ja usein oireellisia, ja jotka liittyivät joihinkin toisen asteen AV-katkoksen vaiheisiin, sekä tyypin 1 että 2, ja joihinkin yöllisen täydellisen av-dissosiaation vaiheisiin. Ekokardiografia ja ergometrinen testi ei paljastanut merkittäviä muutoksia, lukuun ottamatta patenttia foramen ovale. Äidillä ei ollut muita kliinisiä oireita, altistavaa tilaa tai riskitekijöitä. Kliinisen kuvan perusteella probandin äitiin istutettiin PMK: ta. Lisäksi sukuhistoria paljasti, että myös probandin isoäiti ja isoisoisä istutettiin PMK: Hon 55-vuotiaana ja 30-vuotiaana. Lisäksi probandin isoäidin veli kuoli 51-vuotiaana vaikeaan laajentuneeseen kardiomyopatiaan. Kardiologiset tutkimukset tehtiin myös probandin isälle ja sedälle, joiden tulokset olivat täysin ennallaan.
Santa Lucia-säätiön eettinen toimikunta hyväksyi tämän tutkimuksen, ja se tehtiin Helsingin julistuksen mukaisesti. Kaikki osallistujat antoivat allekirjoitetun tietoon perustuvan suostumuksen geneettiseen analyysiin, ja tältä osin he antoivat myös suostumuksen tämän tapausraportin julkaisemiseen.
laboratoriotutkimukset ja diagnostiset testit
genomi-DNA uutettiin perifeerisestä verestä (400 µL) käyttäen MagPurix Blood DNA Extraction Kit-pakettia ja Magpurix Automatic Extraction System (Resnova) – järjestelmää valmistajan ohjeiden mukaisesti. Näytteet sekvensoitiin Ion PGM-järjestelmän ja ioni Ampliseq räätälöity paneeli korkea spesifisyys (Thermo Fisher Scientific). Raadin koko oli 129.13 Kb, jonka odotetaan seulovan ~99,72% koko paneelista vähintään 20x: n kattavuudella. paneeliin kuului 18 geeniä, jotka valittiin tieteellisen kirjallisuuden, Yleisnäkymien (www.ncbi.nlm.nih.gov/books/NBK1116/) ja patogeenisten varianttien esiintymistiheys koko väestössä. Taulukossa 1 on tiivistelmä NGS-paneelin yksityiskohtaisesta kuvauksesta.
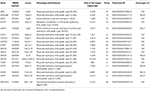
Taulukko 1. Mukautettu NGS paneeli käytetään lgmd diagnoosi.
kirjastojen rakentamisen suoritti Ion AmpliSeq™ Library Kits 2.0. Multiplex PCR-reaktioissa käytettiin noin 10 ng/µl alku-DNA: ta. Peräkkäin, kaksi puhdistusvaihetta (käyttäen AMPure XP, Beckman Coulter) suoritettiin poistaa ei-toivottuja epäpuhtauksia ja lopullinen PCR suoritettiin. Mallin vahvistus-ja rikastusvaiheita suorittivat Ion PGM Hi-Q OT2 kit-400, Ion OneTouch 2 System ja Ion OneTouch ES (Thermo Fisher Scientific). Näytteet käsiteltiin Ion PGM Hi-Q sekvensointi Kit (400 bp, Thermo Fisher Scientific) ja ajaa Ion 316 siru v2 (850 virtaa tarvitaan) ja Ion PGM sekvensseri (Thermo Fisher Scientific). Tulokset analysoitiin Ion Reporter 4.6: n (Thermo Fisher Scientific) ja Integrated Genome Viewerin (IGV) avulla. Geenivarianttien tulkinnasta vastasivat Human Gene Mutation Database (Hgmd), Leiden Open Variation Database (LOVD), ClinVar ja ExAC. Havaittujen muunnosten funktionaalista vaikutusta arvioitiin bioinformatic predictive-työkaluilla, kuten Mutation Taster, Varsome, SIFT, PolyPhen 2, SMART, Human Splicing Finder (HSF). Suora sekvensointi (BigDye Terminator v3.1, BigDyeX Terminator ja ABI3130, Applied Biosystems) tehtiin vahvista geneettisiä variantteja ja genomien sekvenssi koodaus alueiden kattavuus <20X (LMNA, Chr1:156106052-156106076; DYSF, Chr2:71753352-71753502, Chr2:71776385-71776640; SGCB, Chr4:52904225-52904560; SGCA, Chr17:48243242-48243570).
tulokset
probandin NGS-analyysi paljasti 3 heterotsygoottista varianttia (täydentävä Kuva 1). CAPN3: ssa lokalisoitiin kaksi varianttia, nm_000070.2 (CAPN3): c.550delA (p.Thr184Argfs) ja C.1813G>C (P.Val605Leu) eksoneissa 4 ja 16. Ensimmäinen on yksittäinen nukleotidi-deleetio, ja se on yleisin (75% tapauksista) patogeeninen muunnos Euroopan maissa (1). Odotetusti bioinformaattisessa analyysissä C.550delA (rs80338800) luokiteltiin funktion menettäneeksi muunnokseksi (p.Thr184Argfs), mikä aiheutti avoimen lukukehyksen kehyksensiirron. Predictive tools (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) kuvaili muunnosta tuhoisaksi. Lisäksi SMART paljasti, että muutetusta proteiinituotteesta puuttuu calpain 3-domain ja kolme ”EF-hands” – motiivia. Ensimmäinen domeeni osallistuu calpainin signalointireitteihin, kun taas EF-kädet ovat välttämättömiä proteiinin Ca++ – riippuvaiselle aktivaatiolle (4).
koskien c.1813G>C, se on uusi missense-muunnos (s.Val605Leu), jonka on ennustettu olevan haitallinen (mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) mahdollisen jatkomuutoksen vuoksi. Tätä muunnosta ei ole selitetty kirjallisuudessa tai online-tietokannoissa, eikä sitä ole löydetty 200 kontrollihenkilöltä. Valitettavasti SMART Toolin analyysi ei tuottanut merkittäviä tuloksia, koska variantti sijaitsee tuntemattomassa verkkotunnuksessa. CAPN3: n erotteluanalyysi osoitti, että äiti ja eno olivat molemmat heterotsygootteja C.550DELALLE, kun taas isä kantoi C.1813g>C.
lisäksi probandin NGS-analyysi paljasti, että lmna: ssa oli uusi muunnos, nimittäin NM_170707 (Lmna): c.550C>T (P.Gln184*). Ylimainitun muunnoksen on ennustettu olevan null-muunnos, sitä ei ole vielä kuvattu kirjallisuudessa tai nettitietokantojen joukossa eikä sitä ole löydetty 200 kontrollihenkilöltä. Predictive tools (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) kuvasi tämän muunnoksen patogeenisen vaikutuksen proteiinituotteeseen. SMART tool raportoi, että muuttuneesta lmna-proteiinista puuttuu filamenttitunnus, joka on välttämätön sen rakenteen ja toiminnan ylläpitämiseksi. Erotteluanalyysi korosti C: n läsnäoloa.550c>t-muunnos vain äidillä, mikä viittaa mahdolliseen yhteyteen sydämen oireiden ja suvussa esiintyneen sairauden kanssa.
American College of Medical Genetics (ACMG) Standards and Guidelinesin (23) kriteerien mukaan c.1813g>C (CAPN3) voidaan kuvata todennäköiseksi patogeeniseksi muunnokseksi ottaen huomioon, että se sijaitsee kriittisellä alueella, jossa ei ole hyvänlaatuisia variaatioita (PM1); sitä ei esiinny ExAc -, GnomAD-ja 1000 Genomiselaintietokannoissa (PM2); kun otetaan huomioon resessiivinen periytymismalli, se on havaittu transissa, jossa on patogeeninen variantti (PM3); useat laskennalliset todisteet tukevat haitallista vaikutusta geeniin tai geenituotteeseen (PP3). ACMG: n C.550C>t (LMNA) kliinisestä luokituksesta voidaan todeta, että se voidaan nimetä patogeeniseksi muunnokseksi, koska se on null-muunnos, joka aiheuttaa toiminnan menetystä (P. Gln184*) LMNA: ssa (PVS1); sitä ei esiinny ExAc -, GnomAD-ja 1000 Genomiselaintietokannoissa (PM2); se sijaitsee kriittisellä alueella ilman hyvänlaatuista vaihtelua (PM1); useat laskennalliset todisteet tukevat haitallista vaikutusta geeniin tai geenituotteeseen (PP3).
päätelmät
tässä tapausselostuksessa esiteltiin lgmd-tautia sairastava potilas, jonka todettiin kantavan mutaatioita paitsi CAPN3: ssa (c.550delA ja C.1813g>C) myös lmna: ssa (c.550C>T). Nämä tulokset selittävät probandin neuromuskulaarisen fenotyypin CAPN3-mutaatioiden vuoksi ja korostavat mahdollista sydän-ja verisuonisairauksien riskiä, joka johtuu variantin esiintymisestä LMNA: ssa ja hänen positiivisesta suvustaan. Näiden tulosten perusteella perheenjäsenille tehtiin erotteluanalyysi. Emoksi muodostui erityisesti capn3_c.550delA ja LMNA_c.550c>T. Emoa voidaan pitää terveenä kantajana kalpapainopatiaan liittyvälle CAPN3_c.550delA-patogeeniselle mutaatiolle. Lmna: n uuden variantin esiintyminen voi kuitenkin selittää hänen sydän-ja verisuonipatologiansa (bradykardia ja synkopaalijaksot). Äidin neuromuskulaaristen oireiden puuttuminen ja probandin erikoinen kliininen kuva sulkivat pois lmna_c: n mahdollisen yhteyden.550c>T neuromuskulaarisella fenotyypillä, erityisesti Emery-Dreifussin lihasdystrofian (EMD) osalta. Itse asiassa EMD vaikuttaa erityisesti vastus lateralis ja hauis brachii lihaksia, jotka ovat suhteellisen säästynyt lgm2a (24, 25).
eno oli heterotsygoottinen vain capn3_c.550delalle, eikä hänellä odotetusti ollut hermo-tai sydänongelmia. Isä kantoi novellia CAPN3_c.1813G>C, ja näin ollen hän jäi vaille huomiota. Kaiken kaikkiaan erotteluanalyysi vahvisti probandin kolmen mutaation periytymisen hänen sukulaisiltaan ja korosti kardiomyopatian tuntemusta, jota ei voida laiminlyödä (kuva 3).
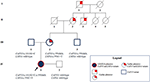
kuva 3. Sukutaulu, joka osoittaa, että äidinpuoleinen perimä periytyy sydämen fenotyypille ja että CAPN3-ja LMNA-muunnokset siirtyvät koko perheenjäseneen.
kaiken kaikkiaan nämä tiedot herättävät joitakin tärkeitä huomioita. Ensinnäkin tämän potilaan NGS-paneeli oli kriittinen kalpapainopatian molekyylidiagnoosin saavuttamiseksi, ja se havaitsi yhden tunnetun mutaation ja toisen uuden muunnoksen, jonka ennustettiin olevan patogeeninen. Toiseksi NGS-paneeli mahdollisti uuden lmna_c.550C>T-muunnoksen tunnistamisen probandissa. Tämä tulos oli yhdenmukainen positiivisen perhehistorian ja eriytymistietojen kanssa, mikä selittää äidin sydän-ja verisuonitauteja ja mikä tärkeintä, suositteli probandin tarkempaa kardiologista seurantaa. On tärkeää huomata, että kardiologiset ilmenemismuodot tapahtuivat äidissä myöhemmin iässä (55 vuotta), jotta voimme olettaa, että proband voisi edelleen olla ”oireettomassa kardiologisessa vaiheessa.”Lmna-mutaation muuttuvaa fenotyyppistä ilmentymistä probandissa verrattuna äitiin ei kuitenkaan voida sulkea pois. Kaikki nämä tiedot korostavat kliinikoiden ja geneetikkojen välisen yhdennetyn lähestymistavan merkitystä tulosten oikean tulkinnan, asianmukaisen geneettisen neuvonnan ja lopulta kliinisen hoidon ja seurannan kannalta. Mitä tulee geneettiseen ja familiaaliseen neuvontaan, NGS-analyysi tehtiin myös probandin kumppanille, jotta voitiin arvioida parille aiheutuva lisääntymisriski. Kumppani oli negatiivinen kaikille 18 testatulle geenille, mikä tarkoittaa, että hänellä on 1/650 jäännösriski olla terve kantaja lgmd: n aiheuttaville mutaatioille, ottaen huomioon, että testi on 84-prosenttisesti herkkä. Kun otetaan huomioon probandin geneettinen profiili ja NGS-testin herkkyys, jäännösriski pariskunnalle saada kalpapainopatiaan sairastunut lapsi on 1/1300. Toisaalta lmna-patogeeniset muunnokset välittyvät autosomaalisen dominanttikuvion mukaan, eli probandilla on 50 prosentin todennäköisyys saada heterotsygootteja lapsia. Jälkeläisten kliinistä kuvaa ei kuitenkaan voida varmasti ennustaa, sillä lmna_c.550c>T on uusi muunnos, eikä sen funktionaalista vaikutusta fenotyyppiin vielä tunneta.
yhteenvetona voidaan todeta, että tässä tapauskertomuksessa korostetaan NGS-paneelien kliinistä hyödyllisyyttä antaa tarkka lgmd2a-diagnoosi ja kuvata monimutkaisia fenotyyppejä ja liitännäissairauksia, jotka johtuvat erilaisten mutaatioiden periytymisestä useissa geeneissä. NGS: n käyttö kliinisessä käytännössä on kuitenkin aina yhdistettävä ennen ja jälkeen geneettisen hoidon, jotta saadaan selkeä selitys tuloksista, mahdollisista vaikutuksista potilaiden fenotyyppiin, uusiutumisriskistä perheen sisällä sekä mahdollisista odottamattomista löydöksistä.
tiedon saatavuus
tätä tutkimusta varten ei luotu tai analysoitu aineistoja.
eettinen lausunto
tämä tutkimus toteutettiin Santa Lucia-säätiön eettisen komitean suositusten mukaisesti kaikkien tutkittavien kirjallisella suostumuksella. Kaikki tutkittavat antoivat kirjallisen tietoon perustuvan suostumuksen Helsingin julistuksen mukaisesti. Pöytäkirjan hyväksyi Santa Lucia-säätiön eettinen komitea.
Tekijäosuudet
RC, CSt, VC, GC, RG, GPa, SC, CP ja JM antoivat panoksensa tiedonhankintaan, analysointiin ja tulkintaan. Käsikirjoituksen laatimiseen ovat osallistuneet CSt, RC, GC, RG, GM ja EG. SZ, GPr, CSa ja SS ovat osallistuneet kliinisen tiedon hankintaan. RC, CSt, VC, GC, RG, GPa, SC, CP, JM, SZ, GPr, GM, CSa, SS ja EG ovat antaneet lopullisen hyväksynnän julkaistavalle versiolle.
Rahoitus
tätä työtä tukee 5x 2016 Kansallinen terveysministeriö 2.
Eturistiriitalausunto
kirjoittajat toteavat, että tutkimus tehtiin ilman kaupallisia tai taloudellisia suhteita, joita voitaisiin pitää mahdollisena eturistiriitana.
Täydennysaineisto
tämän artikkelin Täydennysaineisto löytyy verkosta osoitteesta: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. proteiini ja geneettinen diagnoosi raajan vyö lihasdystrofia tyyppi 2A: tuotto ja sudenkuopat. Lihashermo. (2015) 52:163–73. doi: 10.1002 / mus.24682
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Nigro V, Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol. (2014) 33:1–12.
PubMed Abstract / Google Scholar
3. Rikhard I, Hogrel JY, Stockholm D, Payan CAM, Fougerousse F, Eymard B, et al. Natural history of lgmd2a for detineating outcome measures in clinical trials. Ann Clin Transl Neurol. (2016) 3:248–65. doi: 10.1002 / acn3.287
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Angelini C, Fanin M. Kalpainopatia. Julkaisussa: Adam MP, Ardinger HH, Pagon Ra editors. GeneReviews®. Seattle, WA: University of Washington (2017) 1-30.
PubMed Abstract / Google Scholar
5. Kyriakides T, Angelini C, Schaefer J, Sacconi S, Siciliano G, Vilchez JJ, et al. EFNS: n ohjeet pauci – tai oireettoman hyperckemian diagnostisesta lähestymistavasta. Eur J Neurol. (2010) 17:767–73. doi: 10.1111 / j.1468-1331.2010. 03012.x
CrossRef Full Text | Google Scholar
6. Mori-Yoshimura M, Segawa K, Minami n, Oya Y, Komaki H, Nonaka I, et al. Kardiopulmonaalinen toimintahäiriö potilailla, joilla on raajan vyön lihasdystrofia 2A. Lihashermo. (2017) 55:465–9. doi: 10.1002 / mus.25369
CrossRef Full Text | Google Scholar
7. Okere A, Reddy SS, Gupta S, Shinnar M. kardiomyopatia potilaalla, jolla on raajan vyön lihassurkastumatyyppi 2A. (2013) 6:e12–13. doi: 10.1161 / CIRCHEARTFAILURE.112.971424
CrossRef Full Text | Google Scholar
8. Kramerova I, Ermolova N, Eskin A, Hevener A, Quehenberger O, Armando AM, et al. Epäonnistuminen up-säännellä transkriptio geenien tarpeen lihasten sopeutumista taustalla raajan vyön lihasdystrofia 2A (kalpainopatia). Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093 / hmg/ddw086
PubMed Abstract / CrossRef Full Text | Google Scholar
9. Thompson R, Straub V. Limb girdle muscular dystrophies-international collaborations for translational research. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038 / nrneurol.2016.35
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Strafella C, Caputo V, Galota MR, Zampatti s, Marella G, Mauriello s, et al. Täsmälääketieteen soveltaminen neurodegeneratiivisiin sairauksiin. Etuhermo. (2018) 9:701. doi: 10.3389 / fneur.2018.00701
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Cascella R, Strafella C, Caputo V, Errichiello V, Zampatti s, Milano F, et al. Kohti täsmälääketieteen soveltamista ikään liittyvässä silmänpohjan Rappeumassa. Prog Retin Eye Res. (2017) 63:132-46. doi: 10.1016 / j.preteyeres.2017.11.004
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Cascella R, Strafella C, Longo G, Manzo L, Ragazzo M, De Felici C, et al. Arvioidaan yksilöllistä riskiä AMD kanssa geneettinen neuvonta, suvussa, ja geneettinen testaus. Silmä. (2017) 32:446–50. doi: 10.1038 / silmä.2017.192
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Adams Tri, Eng CM. Seuraavan sukupolven sekvensointi geneettisten häiriöiden diagnosoimiseksi. N Engl J Med. (2019) 379:1353–62. doi: 10.1056 / NEJMc1814955
PubMed Abstract / CrossRef Full Text | Google Scholar
14. Angelini C. neuromuskulaarinen sairaus. Diagnoosi ja löytö raajan vyön lihasdystrofiassa. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038 / nrneurol.2015.230
CrossRef Full Text | Google Scholar
15. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, et al. Käyttö koko-exome sekvensointi diagnoosi raajan vyön lihasdystrofia: tulokset ja saadut kokemukset. JAMA Neurol. (2015) 72:1424–32. doi: 10.1001 / jamaneurol.2015.2274
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Milic A, Daniele N, Lochmüller H, Mora M, Comi GP, Moggio M, et al. Kolmanneksella lgmd2a-koepaloista on normaali calpain 3-proteolyyttinen aktiivisuus in vitro-määrityksellä määritettynä. Neuromusc Disord. (2007) 17:148–56. doi: 10.1016 / j.nmd.2006.11.001
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Lanzillo R, Aurino s, Fanin M, Aguennoz M, Vitale F, Fiorillo C, et al. Varhainen kalpapainopatia normaali ei-toiminnallinen kalpaiini 3 tasolla. Dev Med Child Neurol. (2006) 48:304–6. doi: 10.1017 / S001216220600065X
PubMed Abstract / CrossRef Full Text | Google Scholar
18. Talim B, Ognibene A, Mattioli E, Richard I, Anderson LV, Merlini L. normaali kalpain ilmentymä geneettisesti vahvistetussa raaja-vyön lihassurkastumatyypissä 2A.Neurologia. (2001) 56:692–3. doi: 10.1212 / wnl.56.5.692-a
CrossRef Full Text | Google Scholar
19. Díaz-Manera J, Llauger J, Gallardo E, Illa I. Muscle MRI in muscular dystrophies. Acta Myol. (2015) 34:95–108.
PubMed Abstract / Google Scholar
20. Mercuri E, Bushby K, Ricci E, Birchall DR, Pane M, Kinali M, et al. Lihasten MAGNEETTIKUVAUSLÖYDÖKSET potilailla, joilla on raajan vyön lihasdystrofia, johon liittyy kalpain 3-puutos (LGMD2A) ja varhaisia kontraktuuroja. Neuromusc Disord. (2005) 15:164–71. doi: 10.1016 / j.nmd.2004.10.008
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Magri F, Nigro V, Angelini C, Mongini T, Mora M, Moroni i, et al. Italian raajan vyön lihasdystrofian Rekisteri: suhteellinen esiintymistiheys, kliiniset ominaisuudet ja erotusdiagnoosi. Lihashermo. (2017) 55:55–68. doi: 10.1002 / mus.25192
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Chae J, Minami N, Jin Y, Nakagawa M, Murayama K, Igarashi F, et al. Calpain 3 gene mutations: genetic and clinico-pathologic findings in limb-girdle muscular dystrofy. Neuromusc Disord. (2001) 11:547–55. doi: 10.1016 / S0960-8966(01)00197-3
PubMed Abstrakti / CrossRef kokoteksti / Google Scholar
23. Richards s, Aziz N, Bale s, Bick D, Das s, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038 / gim.2015.30
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Bonne G, Mercuri E, Muchir A, Urtizberea A, Bécane HM, Recan D, et al. Autosomaalisen dominantin Emery-Dreifussin lihasdystrofian kliininen ja molekyyligeneettinen kirjo, joka johtuu lamin A/C-geenin mutaatioista. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170::AID-ANA6>3.0.CO; 2-J
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Hong JS, Ki CS, Kim JW, suh YL, Kim JS, Baek KK, et al. Sydämen rytmihäiriöt, kardiomyopatia ja lihasdystrofia potilailla, joilla on Emery-Dreifussin lihasdystrofia ja raajan vyön lihasdystrofia tyyppi 1B. J Korean Med Sci. (2005) 20:283–90. doi: 10.3346 / jkms.2005.20.2.283
CrossRef Full Text / Google Scholar