- Introduction
- Methods
- ribosomaalisten Profilointitietojen suodatus
- koottavat Virtuaaligenomit
- Ribo-seq-Lukukartoitetun alueen määrittäminen Circrnas
- Rmrj: ien mallin ja luokituksen koulutus
- Rmrjs
- tulokset ja keskustelu
- Translated circRNAs in Humans and A. thaliana
- ihmisen ja A. thaliana circrnasin funktionaalinen rikastus, jonka Koodauspotentiaali on
- Tarkkuuskoe CircCode
- Ribo-seq-tietojen Sekvensointisyvyyden vaikutus
- Circcoden vertailu muihin työkaluihin
- johtopäätökset
- saatavuus ja vaatimukset
- tiedon Saatavuuslausunto
- tekijän osuudet
- Rahoitus
- eturistiriita
- Täydennysaineisto
Introduction
Circrnas (circRNAs) on erityinen Koodaamaton RNA-molekyyli, josta on tullut kuuma tutkimusaihe RNA: n alalla ja joka saa paljon huomiota (Chen and Yang, 2015). Verrattuna perinteiseen lineaariseen RNAs: ään (joka sisältää 5′ ja 3′ päät), circRNA-molekyyleillä on yleensä suljettu pyöreä rakenne; tekee niistä vakaampia ja vähemmän alttiita hajoamiselle (Vicens and Westhof, 2014). Vaikka sirkrnojen olemassaolo on tiedetty jo jonkin aikaa, näitä molekyylejä pidettiin RNA: n sekoittumisen sivutuotteena. Korkean suoritustehon sekvensointi-ja bioinformatiikkateknologioiden kehittymisen myötä circrnat ovat tulleet laajalti tunnetuiksi eläimissä ja kasveissa (Chen and Yang, 2015). Viimeaikaiset tutkimukset ovat myös osoittaneet, että suuri määrä sirkrnoja voidaan muuntaa pieniksi peptideiksi soluissa (Pamudurti et al., 2017)ja ovat avainrooleissa, vaikka niiden joskus alhainen ilmaisutaso (Hsu and Benfey, 2018; Yang et al., 2018). Vaikka sirkrnoja tunnistetaan yhä enemmän, niiden toimintaa kasveissa ja eläimissä on yleensä vielä tutkittava. Mirna-syötteinä toimimisen lisäksi circrnoilla on tärkeä translaatiopotentiaali, mutta työkaluja näiden molekyylien translaatiokyvyn ennustamiseen ei ole (Jakobi and Dieterich, 2019).
sirkrnojen ennustamiseen ja tunnistamiseen on olemassa useita työkaluja, kuten CIRI (Gao et al., 2015), CIRCexplorer (Dong et al., 2019), CircPro (Meng et al., 2017), ja circtools (Jakobi et al., 2018). Niistä CircPro voi paljastaa käännetty circRNAs laskemalla käännös potentiaalinen pisteet circRNAs perustuu CPC (Kong et al., 2007), joka on työkalu avoimen lukukehyksen (ORF) tunnistamiseen tietyssä järjestyksessä. Kuitenkin, koska jotkut circrnat eivät käytä start kodonia käännöksen aikana (Ingolia et al., 2011; Slavoff et al., 2013; Kearse and Wilusz, 2017; Spealman et al., 2018), työllistävät CPC voi suodattaa joitakin todella käännetty circRNAs. Tässä tutkimuksessa käytimme BASiNET (Ito et al., 2018), joka on koneoppimismenetelmiin (random forest ja J48-malli) perustuva RNA-luokittelija. Se aluksi muuntaa tietyn koodaavan RNAs: n (positiivinen data) ja koodaamattoman RNAS: n (negatiivinen data) ja esittää ne monimutkaisina verkkoina; sitten se poimii näiden verkkojen topologiset Mittarit ja rakentaa ominaisuusvektorin kouluttaakseen mallia, jota käytetään luokittelemaan circRNAs: n koodauskapasiteetti. Tällä menetelmällä vältetään virheellinen suodatus käännetty circRNAs, joita ei ole aloitettu AUG. Lisäksi Ribo-seq-tekniikka, joka perustuu korkean suoritustehon sekvensointi seurata rpfs (ribosomaalinen suojattu fragmentit) transkriptien (Guttman et al., 2013; Brar and Weissman, 2015), voidaan käyttää määrittämään sijainnit circRNAs, jotka käännetään (Michel and Baranov, 2013). Tunnistaaksemme circRNAs: n koodauskyvyn kehitimme työkalun CircCode, johon liittyy Python 3–pohjainen kehys, ja sovellimme Circcodea tutkiaksemme circRNAs: n translaatiopotentiaalia ihmisistä ja Arabidopsis thalianasta. Työmme tarjoaa runsaasti resursseja edelleen tutkia toimintoja circRNAs koodauskapasiteettia.
Methods
CircCode kirjoitettiin Python 3 – ohjelmointikielellä; se käyttää Trimmomaattista (Bolger et al., 2014), bowtie (Langmead and Salzberg, 2012) ja STAR (Dobin et al., 2013) suodattaa raakaa Ribo-seq lukee ja kartoittaa nämä suodatetut lukemat genomiin. CircCode tunnistaa sitten Ribo-seq read-mapped alueet circRNAs jotka sisältävät liittymiä. Sen jälkeen, ehdokas kartoitettu sekvenssit circRNAs lajitellaan perustuu luokittajat (J48 malli) osaksi koodaus RNAs ja noncoding RNAS BASiNET. Lopuksi, lyhyet peptidit tuotettu käännös tunnistetaan mahdollisiksi koodausalueiksi circRNAs. Koko prosessi CircCode koostuu viidestä vaiheesta (Kuva 1).
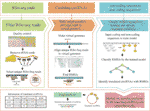
Kuva 1 circcode-työnkulku. Ylin kerros edustaa syöttötiedostoa, joka tarvitaan jokaisessa CircCode – vaiheessa. Keskimmäinen kerros on jaettu kolmeen osaan, ja jokainen osa edustaa eri toimintavaihetta. Vasemmalta oikealle, ensimmäinen osa edustaa suodatus Ribo-seq tiedot; laadunvalvonta suoritetaan Trimmomatic ja rRNA lukee poistetaan bowtie. Toinen osa kuvaa virtuaaligenomin tuottamiseen käytettyjä vaiheita ja kohdentaa suodatetut lukemat virtuaaligenomiin tähdellä. Viimeinen osa edustaa käännettyjen piirien tunnistamista koneoppimisen avulla. Pohjakerros edustaa viimeistä vaihetta, jota käytetään circrnoista käännettyjen peptidien ja lopputuotoksen tulosten ennustamiseen, mukaan lukien tiedot käännetyistä circrnoista ja niiden käännöstuotteista.
ribosomaalisten Profilointitietojen suodatus
ensin heikkolaatuiset fragmentit ja adapterit Ribo-Seq-lukuihin poistetaan Trimmomatiikalla oletusparametreilla, jotta saadaan puhtaat Ribo-seq-lukemat. Toiseksi nämä puhtaat Ribo-seq-lukemat on yhdistetty rRNA-kirjastoon, jotta rRNA: sta johdetut lukemat voidaan poistaa bowtiella. Koska Ribo-seq: n lukupituudet ovat suhteellisen lyhyitä (yleensä alle 50 bp), on mahdollista, että yksi luku vastaa useita alueita. Tällöin on vaikea määritellä, mitä aluetta tietty lukema vastaa. Tämän välttämiseksi puhtaat Ribo-seq-lukemat kartoitetaan kiinnostavan lajin genomiin, ja lukemat, jotka eivät ole täysin linjassa genomin kanssa, katsotaan lopullisiksi ainutlaatuisiksi Ribo-seq-lukemiksi.
koottavat Virtuaaligenomit
Sirkrnat esiintyvät eukaryooteissa yleensä rengasmaisina molekyyleinä, ja ne voidaan tunnistaa niiden selkäliitosten perusteella. Fasta-tiedoston piirien sekvenssit ovat kuitenkin usein lineaarisessa muodossa. Teoriassa tulos osoittaa, että liitos on 5′-terminaalisen nukleotidin ja 3′ – terminaalisen nukleotidin välillä, vaikka liitosta ja sen lähellä olevaa sekvenssiä ei voida tarkastella suoraan, jolloin Ribo-seq lukee circRNA-sekvenssejä, mukaan lukien liitokset, suoraviivaisesti.
CircCode yhdistää kunkin circRNA: n sekvenssin tandemina siten, että kunkin liittymä on keskellä vasta rakennettua sekvenssiä. Erotimme myös jokaisen sarjan yksikön 100 n nukleotidilla, jotta vältetään sekaannus sekvenssin linjausvaiheessa (kunkin RPF: n pituus on alle 50 bp). Lopulta saimme virtuaalisen genomin, joka koostuu vain kandidaattipiireistä, joita erottaa 100 Ns. Koska CircCode keskittyy vain yhdenmukaistaminen Ribo-seq lukee ja circRNA sekvenssejä, voimme tutkia koodaus mahdollisuuksia circRNAs kartoittamalla Ribo-seq lukee tämän virtuaalisen genomin, joka voi säästää paljon laskennallista aikaa (virtuaalinen genomi on paljon pienempi kuin koko genomi) ja lisätä tarkkuutta (välttämällä häiriöitä ylävirran ja loppupään sekvenssivertailut circRNAs).
Ribo-seq-Lukukartoitetun alueen määrittäminen Circrnas
lopulliset ainutlaatuiset Ribo-seq-lukemat on kartoitettu aiemmin luotuun virtuaaliseen genomiin tähden avulla. Koska jokainen tandem circRNA-yksikkö erotettiin 100 n emäksellä ennen virtuaalisen genomin tuottamista, suurin intronin pituus asetettiin enintään 10 emästä parametrilla ” – alignIntronMax 10.”Tämä parametri poistaa kaikki vuorovaikutus eri circRNAs sekvenssin kohdistus. Virtuaaligenomituotannon toisessa vaiheessa CircCode tallentaa sijaintiliittymätiedot jokaiselle virtuaaligenomiin kuuluvalle circRNA: lle. Jos Ribo-seq read-mapped alue virtuaalinen genomi sisältää risteyksen circRNA, ja määrä kartoitettu Ribo-seq lukee junction (NMJ) on suurempi kuin 3, Ribo-seq lukee-mapped alueen junction of circRNAs voidaan pitää rmrj, joka paljastaa karkeasti käännetty segmentti circRNAs lähellä risteyskohtaan.
Rmrj: ien mallin ja luokituksen koulutus
vaikka rmrj: t voivat olla tehokas todiste kääntämisestä, menetelmässä on silti joitakin puutteita. Koska ribosomaalisen kartan lukujen pituus on lyhyt, lukua voidaan verrata väärään kohtaan. Siksi ei ole vakuuttavaa pitää pelkästään Ribo-seq: n kattamaa aluetta käännettynä alueena. Tätä varten koneoppimismenetelmää käytetään rmrj: n koodauskyvyn tunnistamiseen. Ensinnäkin CircCode poimii koodaavia RNAs-koodeja (positiivisia tietoja) ja koodaamattomia RNAS-koodeja (negatiivisia tietoja) kiinnostavasta lajista ja käyttää niitä mallikoulutukseen koodauksen ja koodaamattomien RNAS-koodien välisten ominaisuusvektorien eron avulla. CircCode käyttää koulutettua mallia luokitellakseen Basinetin edellisessä vaiheessa saamat Rmrj: t. Jos sirkrna: n rmrj tunnistetaan koodaavaksi RNA: ksi, tämä sirkrna voidaan tunnistaa käännetyksi sirkrna: ksi.
Rmrjs
kääntyneiden peptidien ennustaminen rmrjs: llä
koska sirkrnojen ilmentyminen eliöissä on vähäistä, Ribo-seq-tiedot eivät osoita tarkkaa 3-nt-jaksotusta selvästi, jos RPFs on pienempi. Siksi on vaikea määrittää tarkka käännös aloittaa sivuston käännetty circRNA. Koska joissakin Rmrj: issä on stop-kodoni ja koska start-kodonia on vaikea määrittää, start-kodoniin ja stop-kodoniin perustuvan ORF-menetelmän löytäminen ei ole mahdollista.
määrittää näiden piirien todelliset käännösalueet ja tuottaa lopullinen käännöstuote, FragGeneScan (Rho et al., 2010), joka pystyy ennustamaan proteiinia koodaavia alueita sirpaloituneissa geeneissä ja geeneissä frameshiftillä, käytetään circrnasin tuottamien käännettyjen peptidien määrittämiseen.
vaivalloisen suoritusprosessin välttämiseksi kaikkia malleja voidaan kutsua komentotulkkikirjoituksella; käyttäjä voi yksinkertaisesti täyttää annetun asetustiedoston ja syöttää sen komentosarjaan, jolloin koko käännettyjen piirien ennustamisprosessi suoritetaan. Lisäksi CircCode voidaan suorittaa erikseen, vaihe vaiheelta siten, että käyttäjä voi säätää parametreja kesken menettelyn ja tarkastella kunkin vaiheen tuloksia halutulla tavalla.
tulokset ja keskustelu
useiden tietokoneiden testauksen jälkeen CircCode todettiin toimivaksi vaadittavien riippuvuuksien ollessa asennettuina. Testaamaan CircCode, käytimme tietoja ihmisille ja A. thaliana ennustaa circRNAs käännöspotentiaalia. Tuloksia verrattiin circrnoihin, jotka on todennettu kokeellisesti vahvistuksena. Sen jälkeen testasimme circcode-koodin false discovery rate (FDR) – arvoa edelleen. Käytimme GenRGenS (Ponty et al., 2006) tuottamaan testausta varten tietokokonaisuuden, joka perustuu tunnettuihin käännettyihin piireihin ja vahvisti, että FDR-arvo oli hyväksyttävällä alueella ja alhaisella tasolla. Lopuksi arvioimme Ribo-seq-datan eri sekvensointisyvyyksien vaikutusta CircCode-ennusteisiin ja vertasimme Circcodea muihin ohjelmistoihin.
Translated circRNAs in Humans and A. thaliana
Soveltaaksemme CircCode-työkalua aitoon dataan latasimme ensin ensembliltä tiedostot, kuten human reference genome GRCh38, genome annotation ja human rRNA. A. thalianan osalta referenssigenomit (TAIR10), genomihuomautustiedostot ja vastaavat rRNA-sekvenssit ladattiin kaikki Ensemblin kasveista. Ribo-seq-tiedot ihmisistä ja A. thalianasta ladattiin rpfdb: stä (liittymisnumerot: GSE96643, GSE81295, GSE88794) (Hsu et al., 2016; Willems et al., 2017), ja kaikki ehdokas circRNAs alkaen human ja A. thaliana ladattiin CIRCPedia v2 (Dong et al., 2018) ja PlantcircBase, vastaavasti (Chu et al., 2017). Lopulta tunnistimme 3,610 käännetty circRNAs ihmisen ja 1,569 käännetty circRNAs alkaen A. thaliana käyttäen CircCode (lisätiedot 1).
ihmisen ja A. thaliana circrnasin funktionaalinen rikastus, jonka Koodauspotentiaali on
käyttäen ihmisen ja A. thalianan CircCode-tuloksia, verkkotyökalua KOBAS 3.0 (Wu et al., 2006) palkattiin merkitsemään nämä käännetyt sirkrnat vanhempien geenien perusteella. Lisäksi suoritimme GO (Gene Ontology) funktionaalianalyysin ja KEGG (Kyoto Encyclopedia of Genes and Genomes) rikastusanalyysin näille käännetyille piireille käyttäen r-pakettia clusterProfiler (Yu et al., 2012).
KEGGIN tulokset osoittivat, että ihmisen sirkrnat rikastuivat proteiinien käsittelyssä endoplasmisessa retikulumireitissä, hiilen metaboloitumisreitissä ja RNA: n kuljetusreitissä. GO-analyysi osoitti ihmisen transloituneiden sirkrnojen osallistumisen molekyylien sitoutumisen, Atpaasiaktiivisuuden ja muiden RNA: n liittämiseen liittyvien biologisten prosessien säätelyyn. Lisäksi A. thalianan käännetyt circrnat rikastuvat stressinkestävyyteen liittyvissä kulkureiteissä, mikä viittaa siihen, että niillä on tärkeä rooli tässä prosessissa (Lisätietoja 2).
Tarkkuuskoe CircCode
circcode-koodin tarkkuuden tutkimiseksi käytettiin Genrgeenien tuottamia testisekvenssejä, joissa käytetään Piilotettua Markovin mallia tuottamaan sekvenssejä, joilla on samat sekvenssiominaisuudet (kuten eri nukleotidien, eri kodonien ja eri nukleotidien taajuudet sekvenssin alussa).
tässä tutkimuksessa käytettiin aiemmin julkaistuja ihmisen kääntämiä sirkrnoja (Yang et al., 2017) syötteenä Genrgensille ja tuotti 10 000 sekvenssiä testaamaan Circcodea. Toistimme testin 10 kertaa, ja keskimäärin 27 käännettyä piiriä ennustettiin joka kerta. FDR-arvoksi laskettiin 0,0027, mikä on paljon vähemmän kuin 0,05, mikä osoittaa, että ennustetut tulokset ovat uskottavia.
lisäksi vertasimme ihmisistä käännettyjä circrnoja, jotka tunnistettiin Circcodella, todennettuihin polysomeihin liittyviin circRNA-tietoihin (Yang et al., 2017). Niistä 60 prosenttia circrnoista tunnistettiin CircCode-koodilla (lisätiedot 3).
Ribo-seq-tietojen Sekvensointisyvyyden vaikutus
selvittääksemme Ribo-seq-tietojen sekvensointisyvyyden vaikutusta CircCode-tunnistustuloksiin testasimme ensin sekvensointisyvyyden vaikutusta käännettyjen piirien määrään (Kuva 2a). Kun sekvensointisyvyys oli pieni, ennustettu käännettyjen piirien määrä oli pieni, ja käännettyjen piirien määrä kasvoi sekvensointisyvyyden kasvaessa. Käännettyjen piirien määrä vakiintui, kun sekvensointisyvyys saavutti peräti 10× lineaarisen transkription kattavuuden.

kuva 2 (A) Ribo-seq-tietojen sekvensointisyvyyden vaikutus käännettyjen piirien ennustettuun määrään. (B) junction read number (JRN): n vaikutus Piirikoodin herkkyyteen eri sekvensointisyvyyksissä.
toiseksi arvioitiin myös NMJ: n vaikutusta herkkyyteen eri sekvensointisyvyyksillä (Kuva 2b). Tulokset osoittivat, että NMJ: llä oli vähemmän vaikutusta herkkyyteen sekvensointisyvyyden kasvaessa. Circcodella oli myös suurempi herkkyys käytettäessä Ribo-seq-dataa suuremmalla sekvensointisyvyydellä.
Circcoden vertailu muihin työkaluihin
Circcoden vertaaminen muihin työkaluihin, kuten Circproon, A. thalianan samaa Ribo-seq-dataa (SRR3495999) käytettiin käännettyjen circrnojen tunnistamiseen kuudella prosessorilla, joissa oli 16 gigatavua RAM-muistia. CircPro tunnisti 44 käännettyä piiriä 13 minuutissa, kun taas CircCode tunnisti 76 käännettyä piiriä 20 minuutissa. Näin ollen CircCode on herkempi kuin CircPro samalla tietokonelaitteistotasolla, mutta se vie enemmän aikaa. CircPro on tiivis ja vähemmän aikaa vievä kuin CircCode, mutta CircCode voi tunnistaa enemmän koodauskykyä omaavia circrnoja kuin CircPro.
johtopäätökset
Sirkrnat ovat tärkeässä asemassa biologiassa, ja on ratkaisevan tärkeää tunnistaa koodauskykyiset sirkrnat tarkasti myöhempää tutkimusta varten. Pohjalta Python 3, kehitimme CircCode, helppokäyttöinen komentorivi työkalu, joka on korkea herkkyys tunnistaa käännetty circRNAs Ribo-Seq lukee suurella tarkkuudella. Circcodella on hyvä suorituskyky sekä kasveissa että eläimissä. Tuleva työ lisää loppupään merkkianalyysin Circcodeen visualisoimalla prosessin jokaisen vaiheen ja optimoimalla ennustuksen tarkkuuden.
saatavuus ja vaatimukset
CircCode on saatavilla osoitteessa https://github.com/PSSUN/CircCode; käyttöjärjestelmät: Linux, ohjelmointikielet: Python 3 ja R; muut vaatimukset: beddools(versio 2.20.0 tai uudempi), bowtie, STAR, Python 3-paketit (Biopython, Pandas, Rpy2), R-paketit (BASiNET, Biostrings). Kaikkien vaadittujen ohjelmistojen asennuspaketit ovat saatavilla CircCode-kotisivulla. Käyttäjien ei tarvitse ladata niitä erikseen. CircCode kotisivu tarjoaa myös yksityiskohtaiset käyttöohjeet viitteeksi. Työkalu on vapaasti käytettävissä. Ei ole rajoituksia käyttöä nonacademics.
tiedon Saatavuuslausunto
kaikki olennaiset tiedot ovat käsikirjoituksessa ja sitä tukevissa tiedostoissa.
tekijän osuudet
käsitteellistäminen: PS, GL. Data Curation: PS, GL. Muodollinen analyysi: PS, GL. Kirjallinen – alkuperäinen luonnos: PS, GL. Kirjoittaminen-arvostelu ja editointi: PS, GL.
Rahoitus
tätä työtä on tuettu Kiinan National Natural Science Foundationin (apurahanumerot 31770333, 31370329 ja 11631012), New Century Excellent Talents in University-ohjelman (NCET-12-0896) ja Keskusyliopistojen Perustutkimusrahastojen (no. GK201403004). Rahoittajilla ei ollut roolia tutkimuksessa, sen suunnittelussa, tiedonkeruussa ja analysoinnissa, julkaisupäätöksessä eikä käsikirjoituksen laatimisessa. Rahoittajilla ei ollut roolia tutkimuksen suunnittelussa, tiedonkeruussa ja analysoinnissa, julkaisupäätöksessä tai käsikirjoituksen laatimisessa.
eturistiriita
kirjoittajat toteavat, että tutkimus tehtiin ilman kaupallisia tai taloudellisia suhteita, joita voitaisiin pitää mahdollisena eturistiriitana.
Täydennysaineisto
tämän artikkelin Täydennysaineisto löytyy verkosta osoitteesta: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
lisätiedot 1 / ennustetun käännetyn sirkrna: n ja lyhyen peptidin sekvenssi.
lisätiedot 2 / GO-rikastuksen ja KEGG-rikastuksen tulokset ihmisille ja Arabidopsis thalianalle.
lisätiedot 3 / ennustettujen käännettyjen piirilevyjen Vertailu validoituihin käännettyihin piirilevyihin.
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: joustava trimmeri illumina-sekvenssitietoihin. Bioinformatiikka 30, 2114-2120. doi: 10.1093 / bioinformatiikka / btu170
PubMed Abstract / CrossRef Full Text | Google Scholar
Brar, G. A., Weissman, J. S. (2015). Ribosomiprofilointi paljastaa proteiinisynteesin mitä, milloin, missä ja miten. Nat. Pastori Mol. Cell Biol. 16, 651–664. doi: 10.1038 / nrm4069
PubMed Abstract / CrossRef Full Text | Google Scholar
Chen, L.-L., Yang, L. (2015). Circrna biogeneesin säätely. RNA Biol. 12, 381–388. doi: 10.1080/15476286.2015.1020271
PubMed Abstrakti / CrossRef kokoteksti / Google Scholar
Chu, Q., Zhang, X., Zhu, X., Liu, C., Mao, L., Ye, C., et al. (2017). PlantcircBase: tietokanta kasvi Pyöreä RNAs. Mol. Tehdas 10, 1126-1128. doi: 10.1016 / J. molp.2017.03.003
PubMed Abstract | CrossRef Full Text | Google Scholar
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). Tähti: ultrafast universal RNA-seq aligner. Bioinformatiikka 29, klo 15-21. doi: 10.1093 / bioinformatiikka / bts635
PubMed Abstract / CrossRef Full Text | Google Scholar
dong, R., Ma, X.-K., Chen, L.-L., Yang, L. (2019). ”Genome-wide annotation of circRNAs and their alternative back-splicing/splicing with CIRCexplorer Pipeline,” in Epitranscriptomics. Toim. Wajapeyee, N., Gupta, R. (New York, NY: Springer New York), 137-149. doi: 10.1007/978-1-4939-8808-2_10
CrossRef kokoteksti / Google Scholar
dong, R., Ma, X.-K., Li, G.-W., Yang, L. (2018). CIRCpedia v2: päivitetty tietokanta kattavaa ympyränmuotoista RNA-merkintää ja ekspression vertailua varten. Genomiikka Proteomiikka Bioinf. 16, 226–233. doi: 10.1016 / j.gpb.2018.08.001
CrossRef Full Text | Google Scholar
Gao, Y., Wang, J., Zhao, F. (2015). CIRI: tehokas ja puolueeton algoritmi de Novon ympyränmuotoiseen RNA-tunnistukseen. Genomibiolia. 16, 4. doi: 10.1186 / s13059-014-0571-3
PubMed Abstrakti / CrossRef kokoteksti / Google Scholar
Guttman, M., Russell, P., Ingolia, N. T., Weissman, J. S., Lander, E. S. (2013). Ribosomiprofilointi tarjoaa näyttöä siitä, että suuret koodaamattomat RNA: t eivät koodaa proteiineja. Selli 154, 240-251. doi: 10.1016 / J.solu.2013.06.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Hsu, P. Y., Benfey, P. N. (2018). Pieni mutta mahtava: funktionaalisia peptidejä, joita pienet orfit koodaavat kasveissa. PROTEOMICS 18, 1700038. doi: 10.1002 / pmic.201700038
CrossRef Full Text / Google Scholar
Hsu, P. Y., Calviello, L., Wu, H.-Y. L., Li, F.-W., Rothfels, C. J., Ohler, U., et al. (2016). Superresoluutioinen ribosomiprofilointi paljastaa arabidopsiksessa sensuroimattomat käännöstapahtumat. Proc. Natl. Acad. Sci. 113, E7126-E7135. doi: 10.1073 / pnas.1614788113
CrossRef Full Text | Google Scholar
Ingolia, N. T., Lareau, L. F., Weissman, J. S. (2011). Hiiren alkion kantasolujen ribosomiprofilointi paljastaa nisäkkäiden proteomien monimutkaisuuden ja dynamiikan. Selli 147, 789-802. doi: 10.1016 / J.solu.2011.10.002
PubMed Abstract | CrossRef Full Text | Google Scholar
Ito, E. A., Katahira, I., Vicente, F. F., da, R., Pereira, L. F. P., Lopes, F. M. (2018). BASiNET-BiologicAl Sequences NETwork: a case study on coding and non-coding RNAS identification. Nucleic Acids Res. 46, E96–e96. doi: 10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093 / bioinformatiikka / bty948
CrossRef Full Text / Google Scholar
Kearse, M. G., Wilusz, J. E. (2017). Non-AUG käännös: uusi alku proteiinisynteesiä eukaryootes. Genes Dev. 31, 1717–1731. doi: 10.1101 / gad.305250.117
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, L., Zhang, Y., Ye, Z.-Q., Liu, X.-Q., Zhao, S.-Q., Wei, L., et al. (2007). CPC: arvioi transkriptien proteiini-koodauspotentiaalia sekvenssiominaisuuksien ja tukivektorikoneen avulla. Nukleiinihapot Res. 35, W345–W349. doi: 10.1093/nar / gkm391
PubMed Abstract / CrossRef Full Text | Google Scholar
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read-linjaus Bowtie 2: n kanssa. Nat. Menetelmät 9, 357-359. doi: 10.1038 / nmeth.1923
PubMed Abstract | CrossRef Full Text | Google Scholar
Meng, X., Chen, Q., Zhang, P., Chen, M. (2017). CircPro: integroitu työkalu sellaisten circrnojen tunnistamiseen, joilla on proteiinikoodauspotentiaalia. Bioinformatiikka 33, 3314-3316. doi: 10.1093 / bioinformatiikka / btx446
PubMed Abstract | CrossRef Full Text | Google Scholar
Michel, A. M., Baranov, P. V. (2013). Ribosomiprofilointi: Hi-Def-monitori proteiinisynteesiä varten genomin laajuisessa mittakaavassa: ribosomiprofilointi. Wiley Interdiscip. RNA 4, 473-490. doi: 10.1002 / wrna.1172
PubMed Abstract | CrossRef Full Text | Google Scholar
Pamudurti, N. R., Bartok, O., Jens, M., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., et al. (2017). Käännös sanalle CircRNAs. Mol. Selli 66, 9-21.e7. doi: 10.1016 / J.molcel.2017.02.021
PubMed Abstract | CrossRef Full Text | Google Scholar
Ponty, Y., Termier, M., Denise, A. (2006). GenRGenS: ohjelmisto tuottaa satunnaisia genomisia sekvenssejä ja rakenteita. Bioinformatiikka 22, 1534-1535. doi: 10.1093 / bioinformatiikka / btl113
PubMed Abstract / CrossRef Full Text | Google Scholar
Rho, M., Tang, H., Ye, Y. (2010). FragGeneScan: ennustavat geenit lyhyissä ja virhealttiissa lukemissa. Nucleic Acids Res. 38, E191–e191. doi: 10.1093/nar / gkq747
PubMed Abstract / CrossRef Full Text | Google Scholar
Slavoff, S. A., Mitchell, A. J., Schwaid, A. G., Cabili, M. N., Ma, J., Levin, J. Z., et al. (2013). Peptidominenlöytö lyhyistä avoimista lukukehyskoodatuista peptideistä ihmissoluissa. Nat. Kemiaa. Biol. 9, 59–64. doi: 10.1038 / nchembio.1120
PubMed Abstract | CrossRef Full Text | Google Scholar
Spealman, P., Naik, A. W., May, G. E., Kuersten, S., Freeberg, L., Murphy, R. F., et al. (2018). Säilöttyjä ei-AUG-urfeja, jotka on paljastettu ribosomin profilointitietojen uudella regressioanalyysillä. Genome Res. 28, 214-222. doi: 10.1101 / gr.221507.117
PubMed Abstract | CrossRef Full Text | Google Scholar
Vicens, Q., Westhof, E. (2014). Pyöreän RNAs: n biogeneesi. Selli 159, 13-14. doi: 10.1016 / J.solu.2014.09.005
PubMed Abstract | CrossRef Full Text | Google Scholar
Willems, P., Ndah, E., Jonckheere, V., Stael, S., tarra, A., Martens, L., et al. (2017). N-terminaalinen proteomics avustaa profilointia tutkimattoman käännöksen initiaatiomaiseman Arabidopsis thaliana. Mol. Solu. Proteomics 16, 1064-1080. doi: 10.1074 / mcp.M116. 066662
PubMed Abstract | CrossRef Full Text | Google Scholar
Wu, J., Mao, X., Cai, T., Luo, J., Wei, L. (2006). KOBAS server: verkkopohjainen alusta automaattiseen merkintöihin ja reittien tunnistamiseen. Nucleic Acids Res. 34, W720-W724. doi: 10.1093 / nar/gkl167
PubMed Abstract / CrossRef Full Text | Google Scholar
Yang, L., Fu, J., Zhou, Y. (2018). Circular RNA: t ja niiden kehittyvät roolit Immuunisäätelyssä. Edessä. Immunol. 9, 2977. doi: 10.3389 / fimmu.2018.02977
PubMed Abstract | CrossRef Full Text | Google Scholar
Yang, Y., Fan, X., Mao, M., Song, X., Wu, P., Zhang, Y., et al. (2017). Laaja käännös Pyöreä RNAs ajaa N6-metyyliadenosiini. Cell Res. 27, 626-641. doi: 10.1038 / cr.2017.31
PubMed Abstract | CrossRef Full Text | Google Scholar
Yu, G., Wang, L.-G., Han, Y., He, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar