Bevezetés
a végtag-öv Izomdisztrófiái (LGMDs) a rendellenességek heterogén csoportját tartalmazzák, amelyet a proximális végtag-öv izmok progresszív pazarlása és gyengesége jellemez (1, 2). Az lgmd-k inter és intrafamilialis variabilitást mutatnak, a nagyon enyhe formáktól a súlyos, korai kezdetű, gyorsan progresszív fenotípusokig (2). Az lgmd-k autoszomális domináns (LGMD1) vagy recesszív (LGMD2) kategóriába sorolhatók. Az első csoport általában felnőtt korban jelentkezik, és nem túl gyakori (<az összes Lgmd 10% – a), míg az utóbbiak gyakoribbak (1:15000) (1). A recesszív formák közül az LGMD2A (vagy calpainopathia) a leggyakoribb LGMD világszerte, amely az összes LGMDs eset körülbelül 30% – át érinti (3). A betegség klinikai jelei közé tartozik a lábujjhegyen járás, a waddling járás, a futás és a lépcsőzés nehézsége, a scapular winging. Gyakori az ízületi kontraktúrák, az Achilles-ín rövidülése, a scoliosis, míg az arc-és nyaki izmok nem érintettek (4). A fiatal betegeknél a tünetmentes HyperCKemia (a CPK normál szintjének 5-80-szorosa) a betegség preklinikai stádiumának tekinthető, és több évig is fennállhat (1, 5). Az izomgyengeség kialakulásának kora általában 15 éves korban jelentkezik, bár előfordulhat korábbi (<12 év) vagy későbbi (>30 év) életkorban. A betegség progressziója az ambuláció elvesztéséhez, légzési elégtelenséghez és a tüdő vitális kapacitásának csökkenéséhez vezethet az előrehaladott stádiumokban (6). A szív érintettségéről (szívritmuszavarok, szívvezetési rendellenességek, bal kamrai ejekciós diszfunkció) csak alkalmanként számoltak be (6, 7).
a calpainopathia diagnózisát megerősíti a patogén mutációk kimutatása a CAPN3-ban (15q15.1) (4), különböző alternatív módon összekapcsolt transzkriptumokat kódolva. A teljes hosszúságú átirat azonban elsősorban az izomszövetben fejeződik ki (8). A kódolt fehérje (CAPN3) a nem lizoszomális Ca++-függő cisztein proteáz család tagja. Az izomban a CAPN3 részt vesz a “sarcomere remodelling” – ben, amely elengedhetetlen az izom alkalmazkodásához és növekedéséhez a funkcionális és anyagcsere igényekhez. A mai napig több mint 490 patogén mutációt írtak le a CAPN3-ban, amelyek többsége egy nukleotid változás (4). A CAPN3 mutációit mitokondriális rendellenességekkel, növekedési kudarccal, fokozott oxidatív stresszel és szarkomér disorganizációval társították, amelyek összességében hozzájárulnak ahhoz, hogy az izom nem képes megtartani a terheléseket, ezáltal myofiber degenerációt és izomvesztést okozva (8). A calpainopathia diagnózisa kihívást jelenthet a genetikai heterogenitás és a klinikai és instrumentális minta nem specifikussága miatt. Valójában az izomgyengeség/atrófia, a hipertrófia/pszeudo-hipertrófia és az ín-kontraktúrák eloszlása nagyon gyakran megoszlik más Lgmd-kkel vagy neuromuszkuláris rendellenességekkel. A calpainopathia izombiopsziás mintája általában nem specifikus is, az enyhe izomrendellenességektől a súlyos dystrophiás változásokig. Ezenkívül az immunhisztokémiai / biokémiai markerek általában nem megbízhatóak: a calpain jel normális lehet még egy nem funkcionális fehérje jelenlétében is, és fordítva, még a calpainopathiától eltérő izomdisztrófiákban is csökkenthető.
ezért a pontos és megbízható eredmények érdekében differenciáldiagnózis elvégzése javasolt. E célból molekuláris genetikai vizsgálati megközelítéseket fejlesztettek ki a calpainopathia diagnózisának megerősítésére, beleértve a multigén panelt, az extenzív genomikai (exome/genome szekvenálás) és az egygénes elemzést (közvetlen szekvenálás) (9). A következő generációs szekvenálás (NGS) megvalósítása hasznos volt a diagnosztikai, prediktív vagy terápiás célokra alkalmazandó informatív adatok előállításához (10-12). Az NGS génpanelek egy adott betegséghez vagy kapcsolódó rendellenességek csoportjához kapcsolódó gének elemzésén alapulnak, amelyeket genetikai és fenotípusos heterogenitás jellemez. Az NGS panelek hasznosak lehetnek kevert vagy összetett fenotípusok kimutatására is, amelyek egynél több genetikai hiba szülőktől való örökléséből származnak (13). Általában azok a komplex fenotípusokat mutató betegek, akik negatívak az egyedi tervezésű diagnosztikai génpanelek ismert mutációival szemben, és széles körű differenciáldiagnózist igényelnek, teljes exome vagy genom szekvenálásra jogosultak. A teljes exome / Genom szekvenálás drágább megközelítés az adatkezelés, az értelmezés és az analitikai költségek szempontjából, de növeli a feltételezett genetikai rendellenesség molekuláris diagnózisának valószínűségét (13). Az Lgmd-k esetében a magas diagnosztikai arány, az optimális lefedettség, az érzékenység és a klinikai vizsgálatok specifikusságának biztosítása érdekében kifejezetten ajánlott a dedikált NGS-panelek használata (9). Az NGS panelek tehát az egyik legjobb rendszert képviselik a differenciáldiagnózis megkönnyítésére, az új okozati mutációk azonosítására és a genotípus-fenotípus összefüggések tisztázására (14, 15).
ebben a kéziratban az LGMD2A által érintett beteg esetéről számoltak be, amelyre az NGS panel alkalmazása nemcsak a calpainopathia diagnózisának megerősítését tette lehetővé (mutációk a CAPN3-ban), hanem egy további, új mutáció azonosítását is az lmna génben, amely dilatált kardiomiopátiával társul. Ezen eredmények alapján az elemzést kiterjesztettük a proband családtagjaira, hogy átfogóbb értelmezést nyújtsunk az analitikai adatoknak a kóros fenotípusokkal kapcsolatban.
eset bemutatása
Proband és rokonai klinikai jellemzése
a proband betegség kialakulását 10 éves korban utalták, amikor a borjú hipertrófia jelenlétét jelentette lábujjhegyen járással, futási nehézségekkel, lépcsőzéssel és felállással. A tünetek lassú, de progresszív romlást mutattak, a proximális felső végtag későbbi bevonásával. 13 éves korában véletlenül felfedezték a CPK magas szintjét (~8000 UI/L), amely soha nem társult a myoglobinuriával. Egymás után a beteg több neuromuszkuláris vizsgálaton esett át különböző speciális központokban. Két izom biopsziát végeztünk, amelyek klasszikus dystrophiás képet mutattak (hipertróf / atrófiás rostok, belső magok, nekrotikus rostok, kötőszövet növekedése) nem specifikus jellemzőkkel. Ahogy az várható volt, az immunkémiai és immunoblot vizsgálatok nem voltak meggyőzőek, ami csak a nekrotikus rostokban mutatott abnormális/csökkent (a laminin esetében is); a dystrophin és a calpain immunblotjai normális jeleket mutattak. A calpain-3 immunoblot azonban ismert, hogy nem teljesen érzékeny az LGMD2A diagnózisra, mivel az esetek 20-30% – ában normál mennyiségű fehérje (1, 16-18) jelenik meg. A klinikai megjelenés miatt (hyperCKmia, borjú hipertrófia/pszeudo-hipertrófia) először kizárták a dystrophinopathiákat. Ezenkívül a DMD (Xp21), FKRP (19q13.32) és DYSF (2P13.2) gének elemzése nem mutatott patogén mutációkat. A beteg 30 éves korában érkezett megfigyelésünkre, axiális és övi érintettséget mutatott mind a felső, mind az alsó végtagban, jelentős kacsázó járással és szárnyas lapockákkal. Az alsó végtagokban a gyengeség kiemelkedő volt a quadriceps és a glutei izmokban, amelyek szignifikánsan ipo-atrófiásak voltak, a “látszólag hipertrófiás” (pszeudo-hipertrófia) vádli izmokkal együtt (1.ábra). Görcsöket, myalgiákat és hullámzást nem figyeltek meg. A teljes pneumológiai (spirometria és poliszomnográfia) és kardiológiai (ökográfia, 24 órás EKG Holter monitorozás, szív MRI, teljes kardiológiai fizikai vizsgálat célzott kórtörténettel és cardiovascularis reflex analízissel) vizsgálatok nem tártak fel jelentős rendellenességet. Kizártuk a savas maltázhiányt is (normál enzimaktivitási vizsgálatok limfocitákban/leukocitákban). Az izommágneses rezonancia képalkotás (MRI) a scapularis öv, a paravertebralis izmok, a comb hátsó rekeszizmai (a sartorius és a gracilis izmok viszonylagos megkímélésével) és a láb (különösen a gastrocnemius medialis és a soleus) kiemelkedő érintettségét mutatta (2.ábra). Az érintettség ezen mintájáról már beszámoltak az irodalomban az MRI képről calpainopathiában (19, 20). Ezenkívül az MRI vizsgálat azt mutatta, hogy a borjúizmok hatékonyan atrofikusak voltak, és a zsír beszivárgása jellemezte, ami izmos “megnagyobbodást” okozott.”A klinikai megjelenés, a megjelenés kora, a progresszió sebessége, az izomgyengeség eloszlása és a proband MRI eredményei teljes mértékben összhangban voltak az LGMD2A diagnózisával, amelyet az irodalomban részletesen leírtak (4, 21, 22).
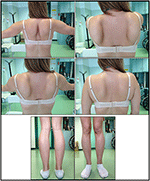
1. ábra. Proband klinikai jellemzők: szárnyas lapocka és comb atrófia és borjú pszeudo-hipertrófia kombinációja.
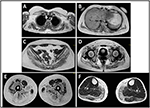
2. ábra. Proband izom MRI: a felső végtagi öv és a paravertebrális izmok (A,B) érintettsége, az alsó végtagi öv és a comb hátsó része (C–E, főleg a glutei) érintettsége a sartorius és a gracilis viszonylagos megkímélésével, valamint a láb hátsó része (F, többnyire gastrocnemius medialis/soleus) érintettsége.
a proband családtagjainak értékelése nem mutatott ki neuromuszkuláris betegséget, és nem mutatott bizonyítékot a szülők közötti rokonságra. A proband anyja azonban 55 éves korában 2 szinkopális epizódot mutatott be, amelyeknél idiopátiás bradycardiát diagnosztizáltak (éjszaka legfeljebb 40 ütés / perc). Kiterjedt kardiológiai vizsgálatok a proband anyján tüneti atrioventrikuláris (AV) blokkot diagnosztizáltak több szinkopális epizód/lipothymia előfordulása után. Az elemzés egy első fokú AV-blokk jelenlétét mutatta ki, több súlyos bradycardia periódussal, többnyire éjszakai és gyakran tüneti, a második fokú AV-blokk egyes fázisaival, mind az 1-es, mind a 2-es típussal, valamint az éjszakai teljes AV-disszociáció néhány fázisával. Az echokardiográfia és az ergometriai vizsgálat nem tárt fel jelentős változásokat, kivéve a foramen ovale szabadalmat. Az anya nem mutatott semmilyen más klinikai tünetet, hajlamosító állapotot vagy kockázati tényezőt. Klinikai képe alapján a proband anyját PMK-val implantálták. Ezenkívül a családtörténetből kiderült, hogy a proband nagymamáját és dédapját is 55, illetve 30 éves korban ültették be PMK-ba. Sőt, a proband nagymamájának testvére 51 éves korában halt meg súlyos dilatált kardiomiopátia miatt. Kardiológiai vizsgálatokat is végeztek a proband apján és anyai nagybátyján, akik teljesen érintetlenek voltak.
ezt a tanulmányt a Santa Lucia Alapítvány etikai bizottsága hagyta jóvá, és a Helsinki Nyilatkozat szerint készült. Valamennyi résztvevő aláírt, tájékozott beleegyezést adott a genetikai elemzéshez, és e tekintetben hozzájárult ezen esetjelentés közzétételéhez is.
laboratóriumi vizsgálatok és diagnosztikai vizsgálatok
genomi DNS-t extraháltunk perifériás vérből (400 6L) MagPurix vér DNS extrakciós készlet és MagPurix automatikus extrakciós rendszer (Resnova) segítségével a gyártó utasításainak megfelelően. A mintákat Ion PGM rendszerrel és Ion Ampliseq Customized Panel High specificitással (Thermo Fisher Scientific) szekvenáltuk. A panel mérete 129 volt.13 Kb, amely várhatóan a teljes panel ~99,72% – át szűri, minimális lefedettséggel 20X .a panel 18 gént tartalmazott, amelyeket tudományos irodalom, általánosítások (www.ncbi.nlm.nih.gov/books/NBK1116/) és a patogén variánsok gyakorisága az általános populációban. Az NGS panel részletes leírását az 1. táblázat foglalja össze.
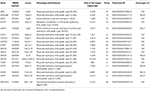
táblázat 1. Testreszabott NGS panel lgmd diagnózishoz.
könyvtárak építési végezte Ion AmpliSeq 6.0 Könyvtár készletek. A multiplex PCR reakciókhoz körülbelül 10 ng / 6L kiindulási DNS-t használtunk. Egymás után két tisztítási lépést (AMPure XP, Beckman Coulter alkalmazásával) végeztünk a nem kívánt szennyeződések eltávolítására, majd egy végső PCR-t végeztünk. A sablonerősítési és dúsítási lépéseket Ion PGM Hi-Q OT2 kit-400, Ion OneTouch 2 Rendszer és Ion OneTouch ES (Thermo Fisher Scientific) végezte. A mintákat Ion PGM Hi-Q szekvenáló készlettel (400 bp, Thermo Fisher Scientific) dolgozták fel, és ION 316 V2 chipen (850 áramlás szükséges) és Ion PGM Szekvenszeren (Thermo Fisher Scientific) futtatták. Az eredményeket Ion Reporter 4.6 (Thermo Fisher Scientific) és Integrated Genome Viewer (IGV) segítségével elemezték. A genetikai variánsok értelmezését a human Gene Mutation Database (HGMD), a Leiden Open Variation Database (LOVD), a ClinVar és az ExAC végezte. A detektált variánsok funkcionális hatását bioinformatikus prediktív eszközökkel értékeltük, beleértve a mutációs kóstolót, Varsome, szitál, PolyPhen 2, intelligens, emberi Splicing Finder (HSF). Közvetlen szekvenálás (BigDye Terminator v3.1, BigDyeX Terminator és ABI3130, Applied Biosystems) genetikai variánsok megerősítésére és a <20x lefedettségű genomi kódoló régiók szekvenálására (Lmna, Chr1:156106052-156106076; DYSF, Chr2:71753352-71753502, Chr2:71776385-71776640; SGCB, Chr4:52904225-52904560; Sgca, chr17:48243242-48243570).
eredmények
a proband NGS analízise 3 heterozigóta variánst tárt fel (1.Kiegészítő ábra). Két variáns lokalizálódott a CAPN3-ban, nevezetesen az NM_000070.2 (CAPN3): c.550dela (p.Thr184Argfs) és c.1813g>C (p.Val605Leu) a 4.és 16. exonban. Az első egy nukleotid deléció, és a leggyakoribb (az esetek 75% – ában) patogén variáns az európai országokban (1). A várakozásoknak megfelelően a bioinformatikai elemzés a c.550delA-t (rs80338800) funkcióvesztés-változatnak (p.Thr184Argfs) minősítette, ami a nyitott olvasási keret kereteltolódását okozta. A prediktív eszközök (mutáció kóstoló, HSF, Varsome, PolyPhen 2, szitál) a változatot károsnak minősítették. Ezenkívül a SMART kiderítette, hogy a megváltozott fehérjetermékből hiányzik a calpain 3 domén és a három “EF-kéz” motívum. Az első domén részt vesz a calpain jelátviteli útvonalaiban, míg az EF-kezek elengedhetetlenek a fehérje Ca++-függő aktiválásához (4).
vonatkozó c.1813G> C, Ez egy új missense variáns (p.Val605Leu), amelyről azt jósolták, hogy káros (mutáció kóstoló, HSF, Varsome, PolyPhen 2, szitál) az illesztés lehetséges megváltozása miatt. Ez a változat nem szerepel a szakirodalomban vagy az online adatbázisokban, és nem található meg 200 kontroll alanyban. Sajnos a SMART tool által végzett elemzés nem eredményezett jelentős eredményeket, mivel a változat egy nem jellemző tartományban található. A capn3 szegregációs elemzése azt mutatta, hogy az anya és az anyai nagybátyja egyaránt heterozigóta volt a c.550dela szempontjából, míg az apa a C.1813g>C.
hordozója volt.ezenkívül a probandon végzett NGS-elemzés egy új variáns jelenlétét tárta fel az LMNA-ban, nevezetesen NM_170707 (LMNA): c.550c>T (p.Gln184*). A fent említett variáns várhatóan nulla variáns, még nem írták le az irodalomban vagy az online adatbázisokban, és 200 kontroll alanyban nem találták meg. Prediktív eszközök (mutációs kóstoló, HSF, Varsome, PolyPhen 2, szitál) leírták ennek a változatnak a fehérjetermékre gyakorolt patogén hatását. A SMART tool arról számolt be, hogy a megváltozott LMNA fehérje hiányzik a filament doménből, ami elengedhetetlen a szerkezetének és funkciójának fenntartásához. A szegregációs elemzés kiemelte a c jelenlétét.550C> T variáns csak az anyában, ami arra utal, hogy lehetséges összefüggés van a szív tüneteivel és a családi kórtörténetével.
az American College of Medical Genetics (ACMG) által megállapított kritériumok szerint szabványok és irányelvek (23), c. 1813g> C (CAPN3) valószínűleg patogén variánsként írható le, tekintve, hogy kritikus tartományban helyezkedik el jóindulatú variáció nélkül (PM1); hiányzik az ExAc, GnomAD és 1000 Genom böngésző adatbázisokban (PM2); figyelembe véve az öröklés recesszív modelljét, transz-ban patogén variánssal (PM3) detektálták; a számítási bizonyítékok több sora támasztja alá a génre vagy a géntermékre (PP3) gyakorolt káros hatást. A C.550c>T (LMNA) klinikai osztályozását illetően az ACMG által patogén variánsként jelölhető meg, mivel ez egy null variáns, amely funkcióvesztést okoz (p.Gln184*) az LMNA-ban (PVS1); hiányzik az ExAc, GnomAD és 1000 Genom böngésző adatbázisokban (PM2); kritikus tartományban helyezkedik el jóindulatú variáció nélkül (PM1); a számítási bizonyítékok több sora támasztja alá a génre vagy a géntermékre (PP3) gyakorolt káros hatást.
következtetések
ez az esetjelentés egy LGMD által érintett beteget mutatott be, akiről kiderült, hogy a mutációk hordozója nemcsak a CAPN3-ban (c.550dela és c.1813g>C), hanem az LMNA-ban is (c.550c> T). Ezek az eredmények magyarázzák a proband neuromuszkuláris fenotípusát a CAPN3 mutációk miatt, és rávilágítanak a cardiovascularis rendellenességek potenciális kockázatára, mivel a variáns jelen van az LMNA-ban és pozitív családi anamnézisében. Ezen eredmények alapján a családtagokat szegregációs elemzésnek vetették alá. Az anya különösen a CAPN3_c.550dela és az LMNA_c.550c>T hordozója lett. Az anya egészséges hordozónak tekinthető a calpainopathiával járó CAPN3_c.550dela patogén mutációban. Azonban egy új változat jelenléte az LMNA-ban megmagyarázhatja a kardiovaszkuláris patológiáját (bradycardia és szinkopális epizódok). A neuromuszkuláris tünetek hiánya az anyában és a proband sajátos klinikai képe kizárta az LMNA_c lehetséges társulását.550c>t neuromuszkuláris fenotípussal, különös tekintettel az Emery-Dreifuss izomdisztrófiára (EMD). Valójában az EMD kifejezetten a vastus lateralis és a bicepsz brachii izmokat érinti, amelyek viszonylag megkíméltek az LGM2A-ban (24, 25).
az anyai nagybátyja csak a CAPN3_c.550dela esetében volt heterozigóta, és a várakozásoknak megfelelően nem mutatott neuromuscularis vagy cardiovascularis problémákat. Az apa eredményezte, hogy készítsen a regény CAPN3_c.1813G>C, és ezáltal nem volt hatással. Összességében a szegregációs elemzés megerősítette a proband három mutációjának öröklődését rokonaitól, és kiemelte a kardiomiopátia ismeretét, amelyet nem lehet elhanyagolni (3.ábra).
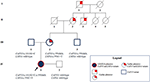
3. ábra. Törzskönyv, amely az anyai származás által örökölt szívfenotípus pozitív ismeretét mutatja, valamint a CAPN3 és az LMNA variánsok átvitelét a családtagok között.
Összességében ezek az adatok néhány fontos szempontot vetnek fel. Először is, az NGS panel ebben a betegben kritikus volt a calpainopathia molekuláris diagnózisának eléréséhez, kimutatva egy ismert mutációt és egy második új variánst, amelyet patogénnek jósoltak. Másodszor, az NGS panel lehetővé tette az új lmna_c.550c>T változat azonosítását a probandban. Ez az eredmény összhangban volt a pozitív családi anamnézissel és a szegregációs adatokkal, megmagyarázva az anya szív-és érrendszeri betegségeit, és ami még fontosabb, specifikusabb kardiológiai nyomon követést javasolt a proband-ban. Fontos megjegyezni, hogy a kardiológiai megnyilvánulások az anyában később (55 év) fordultak elő, így feltételezhetjük, hogy a proband még mindig “tünetmentes kardiológiai fázisban” lehet.”Azonban az LMNA mutáció változó fenotípusos expressziója a probandban az anyához képest nem zárható ki. Mindezek az adatok hangsúlyozzák a klinikusok és a genetikusok közötti integrált megközelítés fontosságát az eredmények helyes értelmezésében, a megfelelő genetikai tanácsadásban és végül a klinikai kezelésben és a nyomon követésben. Ami a genetikai és családi tanácsadást illeti, az NGS elemzést a proband partnerén is elvégezték annak érdekében, hogy megbecsüljék a pár reproduktív kockázatát. A partner negatív volt mind a 18 vizsgált génre, ami azt jelenti, hogy 1/650 maradék kockázata van az LGMD okozta mutációk egészséges hordozójának, figyelembe véve, hogy a teszt 84% – ban érzékeny. Figyelembe véve a proband genetikai profilját és az NGS teszt érzékenységét, a pár fennmaradó kockázata, hogy calpainopathiában szenvedő gyermeket szüljön, 1/1300. Másrészt az LMNA patogén variánsokat autoszomális domináns minta szerint továbbítják, ami azt jelenti, hogy a proband 50% – os valószínűséggel rendelkezik heterozigóta gyermekekkel. Az utódok klinikai képét azonban nem lehet biztosan megjósolni, mivel az LMNA_c.550c>T egy új változat, amelynek funkcionális hatása a fenotípusra még nem ismert.
összefoglalva, ez az esettanulmány kiemeli az NGS panelek klinikai hasznosságát az LGMD2A pontos diagnózisának biztosítására, valamint a különböző gének különböző mutációinak öröklődéséből származó komplex fenotípusok és társbetegségek leírására. Az NGS klinikai gyakorlatban történő alkalmazását azonban mindig kombinálni kell egy pre – és posztgenetikai tanácsadással annak érdekében, hogy egyértelmű magyarázatot adjon az eredményekről, a betegek fenotípusára gyakorolt lehetséges következményekről, a családon belüli kiújulás kockázatáról, valamint a lehetséges váratlan eredmények magyarázatáról.
adatok rendelkezésre állása
ehhez a vizsgálathoz nem hoztak létre vagy elemeztek adatkészleteket.
etikai nyilatkozat
ezt a tanulmányt a Santa Lucia Alapítvány etikai bizottságának ajánlásaival összhangban, minden alany írásbeli tájékozott beleegyezésével végezték. Minden alany írásbeli tájékozott beleegyezést adott a Helsinki Nyilatkozatnak megfelelően. A protokollt a Santa Lucia Alapítvány etikai bizottsága hagyta jóvá.
szerzői hozzájárulások
az RC, CSt, VC, GC, RG, GPa, SC, CP és JM hozzájárult az adatok megszerzéséhez, elemzéséhez és értelmezéséhez. CSt, RC, GC, RG, GM, EG részt vettek a kézirat elkészítésében. Az SZ, a GPr, a CSa és az SS részt vett a klinikai adatok megszerzésében. Az RC, CSt, VC, GC, RG, GPa, SC, CP, JM, SZ, GPr, GM, CSa, SS és EG végleges jóváhagyást adott a közzéteendő verzióra.
finanszírozás
ezt a munkát az 5x 2016 Nemzeti Egészségügyi Minisztérium támogatja 2.
összeférhetetlenségi nyilatkozat
a szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségnek tekinthetők.
Kiegészítő anyag
a cikk kiegészítő anyaga megtalálható az interneten:: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. A 2A típusú végtagöv izomdisztrófia fehérje és genetikai diagnózisa: a hozam és a buktatók. Izom Ideg. (2015) 52:163–73. doi: 10.1002 / mus.24682
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós
2. Nigro V, Savarese M. a végtag-öv izomdisztrófiáinak genetikai alapja: a 2014.évi frissítés. Acta Miol. (2014) 33:1–12.
PubMed Absztrakt / Google Scholar
3. I. Richárd, Hogrel JY, Stockholm D, Payan CAM, Fougerousse F, Eymard B és mtsai. Az LGMD2A természetrajza a klinikai vizsgálatok kimenetelének meghatározására. Ann Clin Transl Neurol. (2016) 3:248–65. doi: 10.1002 / acn3.287
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós
4. Angelini C, Fanin M. Calpainopathia. Ban ben: Adam MP, Ardinger HH, Pagon RA szerkesztők. GeneReviews++. Seattle, WA: Washingtoni Egyetem (2017) 1-30.
PubMed Absztrakt / Google Scholar
5. Kyriakides T, Angelini C, Schaefer J, Sacconi S, szicíliai G, Vilchez JJ, et al. EFNS iránymutatások a pauci – vagy tünetmentes hiperckémia diagnosztikai megközelítéséről. Eur J Neurol. (2010) 17:767–73. doi: 10.1111 / j. 1468-1331.2010. 03012.x
CrossRef teljes szöveg / Google Scholar
6. Mori-Josimura M, Segawa K, Minami N, Oya Y, Komaki H, Nonaka i, et al. Cardiopulmonalis diszfunkció végtag-öv izomdisztrófiában szenvedő betegeknél 2A. Izomideg. (2017) 55:465–9. doi: 10.1002 / mus.25369
CrossRef Teljes Szöveg / Google Scholar
7. Okere a, Reddy SS,Gupta S, Shinnar M. a kardiomiopátia 2A típusú végtagöv izomdisztrófiában szenvedő betegnél. (2013) 6: e12–13. doi: 10.1161 / CIRCHEARTFAILURE.112.971424
CrossRef Teljes Szöveg / Google Scholar
8. Kramerova I, Ermolova N, Eskin A, Hevener A, Quehenberger O, Armando AM, et al. Az izomadaptációhoz szükséges gének transzkripciójának szabályozásának elmulasztása a végtagöv izomdisztrófiájának 2A (calpainopathia) alapja. Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093 / hmg / ddw086
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
9. Thompson R, Straub V. Végtagöv izomdisztrófiák-nemzetközi együttműködések a transzlációs kutatáshoz. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038 / nrneurol.2016.35
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
10. Strafella C, Caputo V, Galota MR, Zampatti S, Marella G, Mauriello S és mtsai. Precíziós gyógyszer alkalmazása neurodegeneratív betegségekben. Elülső Neurol. (2018) 9:701. doi: 10.3389 / fneur.2018.00701
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
11. Cascella R, Strafella C, Caputo V, Errichiello V, Zampatti S, Milano F, et al. A precíziós gyógyszer alkalmazása felé az életkorral összefüggő makula degenerációban. Prog Retin Eye Res. (2017) 63:132-46. doi: 10.1016 / j. preteyeres.2017.11.004
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
12. Cascella R, Strafella C, Longo G, Manzo L, Ragazzo M, De Felici C és mtsai. Az AMD egyéni kockázatának felmérése genetikai tanácsadással, családi anamnézissel és genetikai teszteléssel. Szem. (2017) 32:446–50. doi: 10.1038 / szem.2017.192
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
13. Adams DR, Eng CM. Következő generációs szekvenálás a feltételezett genetikai rendellenességek diagnosztizálására. N Engl J Med. (2019) 379:1353–62. doi: 10.1056 / NEJMc1814955
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
14. Angelini C. neuromuszkuláris betegség. Diagnózis és felfedezés a végtagöv izomdisztrófiájában. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038 / nrneurol.2015.230
CrossRef Teljes Szöveg / Google Scholar
15. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, et al. A teljes exome szekvenálás alkalmazása a végtag-öv izomdisztrófia diagnosztizálására: eredmények és tanulságok. JAMA Neurol. (2015) 72:1424–32. doi: 10.1001 / jamaneurol.2015.2274
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
16. Milic A, Daniele N, Lochm H, Mora M, Comi GP, Moggio M és mtsai. Az LGMD2A biopsziák egyharmadának normális calpain 3 proteolitikus aktivitása van, amelyet in vitro vizsgálat határoz meg. Neuromusc Disord. (2007) 17:148–56. doi: 10.1016 / j. nmd.2006.11.001
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
17. Lanzillo R, Aurino S, Fanin M, Aguennoz M, Vitale F, Fiorillo C és mtsai. Korai kezdetű calpainopathia normál nem funkcionális calpain 3 szinttel. Dev Med Gyermek Neurol. (2006) 48:304–6. doi: 10.1017 / S001216220600065X
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
18. Talim B, Ogribene a, Mattioli E, Richard I, Anderson LV, Merlini L. normál calpain expresszió genetikailag megerősített végtag-öv izomdisztrófiában 2A típusú. neurológia. (2001) 56:692–3. doi: 10.1212 / wnl.56.5.692-a
CrossRef teljes szöveg / Google Tudós
19. D 6-Manera J, Llauger J, Gallardo E, Illa I. izom MRI izomdisztrófiákban. Acta Miol. (2015) 34:95–108.
PubMed Absztrakt / Google Scholar
20. Mercuri E, Bushby K, Ricci E, Birchall DR, ablaktábla M, Kinali M, et al. Izom MRI eredmények calpain 3 hiányban (LGMD2A) szenvedő végtagöv izomdisztrófiában és korai kontraktúrákban szenvedő betegeknél. Neuromusc Disord. (2005) 15:164–71. doi: 10.1016 / j. nmd.2004.10.008
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
21. Magri F, Nigro V, Angelini C, Mongini T, Mora M, Moróni I, et al. Az olasz végtag öv izomsorvadás registry: relatív gyakoriság, klinikai jellemzők és differenciáldiagnózis. Izom Ideg. (2017) 55:55–68. doi: 10.1002 / mus.25192
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós
22. Chae J, Minami N, Jin Y, Nakagawa M, Murayama K, Igarashi F, et al. Calpain 3 génmutációk: genetikai és klinikai-patológiai leletek a végtag-öv izomdisztrófiájában. Neuromusc Disord. (2001) 11:547–55. doi: 10.1016 / S0960-8966(01)00197-3
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
23. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Szabványok és iránymutatások a szekvenciaváltozatok értelmezéséhez: az American College of Medical Genetics and Genomics és az Association for Molecular Pathology közös konszenzusos ajánlása. Genet Med. (2015) 17:405–24. doi: 10.1038 / gim.2015.30
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
24. Bonne G, Mercuri E, Muchir A, Urtizberea A, B Inconcane HM, Recan D, et al. Az autoszomális domináns Emery-Dreifuss izomdisztrófia klinikai és molekuláris genetikai spektruma a lamin A / C gén mutációi miatt. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170::AID-ANA6>3.0.CO; 2-J
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
25. Hong JS, Ki CS, Kim JW, Suh YL, Kim JS, Baek KK, et al. Cardialis dysrhythmiák, cardiomyopathia és izomsorvadás 1b. J típusú Emery-Dreifuss izomsorvadásban és végtag-öv izomsorvadásban szenvedő betegeknél. (2005) 20:283–90. doi: 10.3346 / jkms.2005.20.2.283
CrossRef Teljes Szöveg / Google Scholar