- Bevezetés
- módszerek
- riboszomális profilozási adatok szűrése
- virtuális genomok összeállítása
- a Ribo-seq Read-leképezett Régió meghatározása a Circrns-ek csomópontján (rmrj)
- az Rmrj-K modelljének és osztályozásának képzése
- a lefordított peptidek előrejelzése RMRJs által
- Results and Discussion
- lefordított circrns emberben és A. thaliana
- humán és A. thaliana circrns funkcionális dúsítása
- Circcode pontossági teszt
- a Ribo-seq Adatszekvenálási mélység hatása
- a CircCode összehasonlítása más eszközökkel
- következtetések
- elérhetőség és követelmények
- adatok rendelkezésre állásáról szóló nyilatkozat
- szerzői hozzájárulások
- finanszírozás
- összeférhetetlenség
- Kiegészítő anyag
Bevezetés
a körkörös RNS-ek (circrns-ek) a nem kódoló RNS-molekulák egy speciális típusa, amely az RNS területén forró kutatási témává vált, és nagy figyelmet kap (Chen and Yang, 2015). Összehasonlítva a hagyományos lineáris RNS-ekkel (amelyek 5′ és 3′ végeket tartalmaznak), a circrns-molekulák általában zárt kör alakúak; stabilabbá és kevésbé hajlamosak a lebomlásra (Vicens and Westhof, 2014). Bár a circrns-ek létezése egy ideje ismert, ezeket a molekulákat az RNS splicing melléktermékének tekintették. A nagy áteresztőképességű szekvenálás és a bioinformatikai technológiák fejlesztésével azonban a circrns-ek széles körben elismertek az állatokban és növényekben (Chen and Yang, 2015). A legújabb tanulmányok azt is kimutatták, hogy nagyszámú circrns-t lehet lefordítani kis peptidekké a sejtekben (Pamudurti et al., 2017), és kulcsszerepük van annak ellenére, hogy néha alacsony kifejezési szintjük van (Hsu and Benfey, 2018; Yang et al., 2018). Bár egyre több circrns-t azonosítanak, a növényekben és állatokban betöltött funkcióikat általában még tanulmányozni kell. A miRNS-csaliként betöltött funkcióik mellett a circrns-ek fontos transzlációs potenciállal rendelkeznek, de nem állnak rendelkezésre eszközök ezen molekulák transzlációs képességeinek konkrét előrejelzésére (Jakobi and Dieterich, 2019).
számos eszköz létezik a cirrns-ek előrejelzésére és azonosítására, mint például a CIRI (Gao et al., 2015), CIRCexplorer (Dong et al., 2019), CircPro (Meng et al., 2017) és circtools (Jakobi et al., 2018). Közülük a CircPro felfedheti a lefordított circrns-eket azáltal, hogy kiszámítja a circrns-ek fordítási potenciálját a CPC alapján (Kong et al., 2007), amely eszköz az open reading frame (ORF) azonosításához egy adott sorrendben. Mivel azonban egyes circrns-ek nem használják a start kodont a fordítás során (Ingolia et al., 2011; Slavoff et al., 2013; Kearse and Wilusz, 2017; Spealman et al., 2018), a CPC alkalmazása kiszűrhet néhány valóban lefordított circrns-t. Ebben a tanulmányban a BASiNET-et használtuk (Ito et al., 2018), amely a gépi tanulási módszereken alapuló RNS osztályozó (random forest és J48 modell). Kezdetben átalakítja az adott kódoló RNS-eket (pozitív adatok) és nem kódoló RNS-eket (negatív adatok), és komplex hálózatokként jeleníti meg őket; ezután kivonja ezeknek a hálózatoknak a topológiai méréseit, és létrehoz egy jellemzővektort a circrns-ek kódolási kapacitásának osztályozására használt modell betanításához. Ezzel a módszerrel elkerülhető az Aug által nem kezdeményezett lefordított circrns-ek hibás szűrése. Ezenkívül a Ribo-seq technológia, amely nagy áteresztőképességű szekvenáláson alapul az átiratok RPF-jeinek (riboszomális védett fragmensek) monitorozására (Guttman et al., 2013; Brar and Weissman, 2015), felhasználható a lefordított circrns-ek helyének meghatározására (Michel and Baranov, 2013). A circrns-ek kódolási képességének azonosítására kifejlesztettük a CircCode eszközt, amely egy Python 3–alapú keretrendszert tartalmaz, és a CircCode-ot alkalmaztuk a circrns-ek fordítási potenciáljának vizsgálatára az emberekből és az Arabidopsis thaliana-ból. Munkánk gazdag forrást biztosít a kódolási kapacitással rendelkező circrns-ek funkcióinak további tanulmányozásához.
módszerek
CircCode írták a Python 3 programozási nyelv; használ Trimmomatic (Bolger et al., 2014), bowtie (Langmead and Salzberg, 2012) és STAR (Dobin et al., 2013) a nyers Ribo-seq olvasmányok szűrésére és ezeknek a szűrt olvasatoknak a genomba történő leképezésére. A CircCode ezután azonosítja a Ribo-seq olvasási leképezett régiókat a circrns-ekben, amelyek csomópontokat tartalmaznak. Ezt követően a jelölt leképezett szekvenciákat a circrns-ekben osztályozók (J48 modell) alapján osztályozzák kódoló RNS-ekbe, és nem kódoló RNS-ekbe a BASiNET által. Végül a transzlációval előállított rövid peptideket a circrns-ek potenciális kódoló régióiként azonosítják. A Circode teljes folyamata öt lépésből áll (1.ábra).
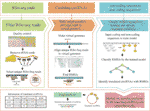
ábra 1 A CircCode munkafolyamata. A felső réteg a CircCode minden lépéséhez szükséges bemeneti fájlt jelöli. A középső réteg három részre oszlik, és mindegyik rész egy másik működési fázist képvisel. Balról jobbra az első rész a Ribo-seq adatok szűrését jelenti; a minőségellenőrzést a Trimmomatic hajtja végre, az rRNS leolvasásokat pedig a bowtie távolítja el. A második rész a virtuális Genom előállításához használt lépéseket mutatja be, és a szűrt leolvasásokat a virtuális genomhoz igazítja a csillaggal. Az utolsó rész a lefordított circrns-ek gépi tanulással történő azonosítását jelenti. Az alsó réteg az utolsó lépés, amelyet a circrns-ekből lefordított peptidek és a végső kimeneti eredmények előrejelzésére használnak, beleértve a lefordított circrns-ekre és azok fordítási termékeire vonatkozó információkat.
riboszomális profilozási adatok szűrése
először is, a Ribo-Seq olvasmányok gyenge minőségű töredékeit és adaptereit a Trimmomatic eltávolítja az alapértelmezett paraméterekkel, hogy tiszta Ribo-seq olvasatokat kapjon. Másodszor, ezek a tiszta Ribo-seq olvasmányok egy rRNA könyvtárba vannak leképezve, hogy eltávolítsák az rRNS-ből származó olvasásokat bowtie segítségével. Mivel a Ribo-seq olvasási hossza viszonylag rövid (általában kevesebb, mint 50 bp), lehetséges, hogy egy olvasás több régiónak felel meg. Ebben az esetben nehéz meghatározni, hogy melyik régiónak felel meg egy adott olvasás. Ennek elkerülése érdekében a tiszta Ribo-seq leolvasásokat leképezik egy érdekes faj genomjához, és azokat az olvasásokat, amelyek nem tökéletesen illeszkednek a genomhoz, a végső egyedi Ribo-seq olvasásnak tekintik.
virtuális genomok összeállítása
a Cirkrns-ek általában gyűrű alakú molekulákként jelennek meg az eukariótákban, és a visszailleszkedő csomópontjaik alapján azonosíthatók. A fasta fájlban található circrns-ek szekvenciái azonban gyakran lineáris formában vannak. Elméletileg az eredmény azt jelzi, hogy a csomópont az 5′ terminális nukleotid és a 3′ terminális nukleotid között van, bár a csomópontot és a csomópont közelében lévő szekvenciát nem lehet közvetlenül megtekinteni, így a Ribo-seq olvasásokat a circrns szekvenciákhoz igazítja, beleértve a csomópontokat is, egyenes módon.
a CircCode összekapcsolja az egyes circrns-ek sorrendjét úgy, hogy mindegyik csomópontja az újonnan felépített szekvencia közepén legyen. Az egyes sorozategységeket 100 N nukleotiddal is elválasztottuk, hogy elkerüljük a zavart a szekvencia-igazítási lépésben (az egyes RPF hossza kevesebb, mint 50 bp). Végül egy virtuális genomot kaptunk, amely csak jelölt circrns-ekből áll, tandemben, 100 Ns-szel elválasztva. Mivel a CircCode csak a Ribo-seq olvasások és a circrns szekvenciák összehangolására összpontosít, megvizsgálhatjuk a circrns-ek kódolási potenciálját a Ribo-seq olvasások ehhez a virtuális genomhoz történő leképezésével, ami nagy mennyiségű számítási időt takaríthat meg (a virtuális Genom sokkal kisebb, mint a teljes genom), és növelheti a pontosságot (elkerülve a circrns-ek upstream és downstream szekvenciájának összehasonlítását).
a Ribo-seq Read-leképezett Régió meghatározása a Circrns-ek csomópontján (rmrj)
a végső egyedi Ribo-seq olvasásokat egy korábban létrehozott virtuális genomhoz térképezzük fel csillag. Mivel minden tandem circrns egységet 100 N bázis választ el egymástól a virtuális Genom előállítása előtt, a legnagyobb intron hosszát úgy állítottuk be, hogy ne haladja meg a 10 bázist a “–alignIntronMax 10 paraméterrel.”Ez a paraméter kiküszöböli a különböző circrns-ek közötti kölcsönhatásokat a szekvencia igazításában. A virtuális genomtermelés második lépésében a CircCode a virtuális genom minden egyes circrns-jére pozicionális csomópont-információkat tárol. Ha a Ribo-seq read-leképezett régió a virtuális genomban tartalmazza a circrns csomópontját, és a leképezett Ribo-seq olvasások száma a csomóponton (NMJ) nagyobb, mint 3, akkor a Ribo-seq reads-leképezett régió a circrns csomópontján RMRJ-nek tekinthető, amely a circrns-ek nagyjából lefordított szegmensét tárja fel a csomópont közelében.
az Rmrj-K modelljének és osztályozásának képzése
bár az Rmrj-k a fordítás hatékony bizonyítékai lehetnek, még mindig vannak hiányosságok ebben a módszerben. Mivel a riboszomális térkép leolvasásainak hossza rövid, az olvasást összehasonlíthatjuk a rossz pozícióval. Ezért nem meggyőző, ha egyszerűen a Ribo-seq által lefedett régiót tekintjük lefordított régiónak. Ebből a célból a gépi tanulási módszert használják az RMRJ kódolási képességének azonosítására. Először is, a CircCode kivonja a kódoló RNS-eket (pozitív adatok) és a nem kódoló RNS-eket (negatív adatok) egy érdekes fajból, és felhasználja őket modellképzésre a kódoló és a nem kódoló RNS-ek közötti jellemző Vektorok különbsége révén. A CircCode ezután a betanított modellt használja az előző lépésben a BASiNET által kapott Rmrj-k osztályozására. Ha egy circrns RMRJ-jét kódoló RNS-ként ismerjük fel, akkor ez a circrns lefordított circrns-ként azonosítható.
a lefordított peptidek előrejelzése RMRJs által
mivel a circrns-ek expressziója organizmusokban alacsony, a Ribo-seq adatok kevesebb RPF esetén nem mutatják egyértelműen a pontos 3-nt periodicitást. Ezért nehéz meghatározni a lefordított circrns pontos fordítási kezdő helyét. Néhány rmrj-ben található stop kodon jelenléte miatt, és mivel a start kodont nehéz meghatározni, a start kodonon és a stop kodonon alapuló ORF megtalálásának módszere nem kivitelezhető.
ezeknek a circrns-eknek a valódi fordítási régióinak meghatározásához és a végső fordítási termék előállításához FragGeneScan (Rho et al., 2010), amely képes megjósolni a fragmentált génekben és a kereteltolódású génekben a fehérjét kódoló régiókat, a circrns-ek által termelt transzlált peptidek meghatározására szolgál.
a nehézkes futási folyamat elkerülése érdekében az összes modell meghívható egy shell szkript segítségével; a felhasználó egyszerűen kitöltheti az adott konfigurációs fájlt, és beírhatja a szkriptbe, majd a lefordított circrns-ek előrejelzésének teljes folyamata lefut. Ezenkívül a CircCode külön is futtatható, lépésről lépésre, így a felhasználó beállíthatja a paramétereket az eljárás közepén, és megtekintheti az egyes lépések eredményeit a kívánt módon.
Results and Discussion
több számítógépen végzett tesztelés után kiderült, hogy a Circode sikeresen fut a szükséges függőségek telepítésével. A CircCode teljesítményének teszteléséhez az emberekre és az A. thaliana-ra vonatkozó adatokat használtuk a transzlációs potenciállal rendelkező circrns-ek előrejelzésére. Az eredményeket összehasonlítottuk azokkal a circrns-ekkel, amelyeket kísérletileg megerősítésként igazoltak. Ezt követően tovább teszteltük a circcode false discovery rate (FDR) értékét. Mi használt GenRGenS (Ponty et al., 2006), hogy az ismert lefordított circrns-eken alapuló vizsgálathoz adatkészletet állítsanak elő, és megerősítsék, hogy az FDR-érték elfogadható tartományon belül és alacsony szinten van. Végül értékeltük a Ribo-seq adatok különböző szekvenálási mélységeinek hatását a CircCode előrejelzésekre, és összehasonlítottuk a CircCode-ot más szoftverekkel.
lefordított circrns emberben és A. thaliana
a CircCode eszköz valós adatokra történő alkalmazásához először letöltöttük a fájlokat, beleértve a grch38 humán referencia genomot, a genome annotation-t és az emberi rRNS-t az Ensembl-ből. Az A. thaliana esetében a referencia genomokat (tair10), a genom annotációs fájlokat és a megfelelő rRNS szekvenciákat mind az Ensembl növényekből töltötték le. Az emberekre és az A. thalianára vonatkozó Ribo-seq adatokat az RPFdb-ről töltötték le (csatlakozási számok: GSE96643, GSE81295, GSE88794) (Hsu et al., 2016; Willems et al., 2017), és az összes jelölt circrns a human és A. a thaliana – t a CIRCPedia v2-ből töltötték le (Dong et al., 2018), illetve a PlantcircBase (Chu et al., 2017). Végül 3610 lefordított circrns-t azonosítottunk emberből és 1569 lefordított circrns-t az A. thaliana-ból CircCode segítségével (kiegészítő adatok 1).
humán és A. thaliana circrns funkcionális dúsítása
kódolási potenciállal az emberi és A. thaliana CircCode eredményeinek felhasználásával, a KOBAS 3.0 online eszköz (Wu et al., 2006) alkalmazták ezeket a lefordított circrns-eket a szülő génjeik alapján. Ezenkívül GO (Gene Ontology) funkcionális elemzést és KEGG (Kyoto Encyclopedia of Genes and Genomes) dúsítási elemzést végeztünk ezekre a lefordított circrns-ekre az R csomag clusterProfiler (Yu et al., 2012).
a KEGG eredmények azt mutatták, hogy az emberi circrns-ek dúsultak a fehérje feldolgozásában az endoplazmatikus retikulum útvonalon, a szén-anyagcsere útvonalon és az RNS transzport útvonalon. A GO analízis kimutatta a humán transzlált circrns-ek részvételét a molekulakötés, az ATPáz aktivitás és más RNS splicinghez kapcsolódó biológiai folyamatok szabályozásában. Ezenkívül az A. thaliana lefordított circrns-jei a stressz-rezisztenciával kapcsolatos útvonalakkal gazdagodnak, ami arra utal, hogy létfontosságú szerepet játszanak ebben a folyamatban (2.Kiegészítő adatok).
Circcode pontossági teszt
a CircCode pontosságának vizsgálatához a genrgens által generált tesztsorozatokat használtuk, amelyek a rejtett Markov modellt használják olyan szekvenciák előállítására, amelyek azonos szekvenciajellemzőkkel rendelkeznek (például különböző nukleotidok, különböző kodonok és különböző nukleotidok frekvenciái a szekvencia kezdetén).
ehhez a tanulmányhoz korábban publikált emberi fordítású circrns-eket használtunk (Yang et al., 2017) A GenRGenS bemeneteként 10 000 szekvenciát generált a CircCode teszteléséhez. 10-szer ismételtük meg a tesztet, és átlagosan 27 fordított circrns-t jósoltunk meg minden alkalommal. Az FDR értékét 0,0027-re számították, ami jóval kevesebb, mint 0,05, ami azt jelzi, hogy az előrejelzett eredmények hitelesek.
ezenkívül összehasonlítottuk a circcode által azonosított emberekből származó lefordított circrns-eket ellenőrzött poliszómához kapcsolódó circrns-adatokkal (Yang et al., 2017). Közülük a circrns-EK 60% – át CircCode azonosította (kiegészítő adatok 3).
a Ribo-seq Adatszekvenálási mélység hatása
a Ribo-seq adatok szekvenálási mélységének a CircCode azonosítási eredményekre gyakorolt hatásának vizsgálatához először teszteltük a szekvenálási mélység hatását a lefordított circrns-ek számára (2a.ábra). Amikor a szekvenálási mélység alacsony volt, a lefordított circrns-ek becsült száma alacsony volt, a lefordított circrns-ek száma pedig a szekvenálási mélység növekedésével nőtt. A lefordított circrns-ek száma stabilvá vált, amikor a szekvenálási mélység elérte a 10-et.

2. ábra (A) A Ribo-seq adatszekvenálási mélység hatása a lefordított circrns-ek becsült számára. (B) a junction read number (JRN) hatása a Circode érzékenységre különböző szekvenálási mélységekben.
másodszor, az NMJ érzékenységre gyakorolt hatását különböző szekvenálási mélységekben is értékeltük (2b ábra). Az eredmények azt mutatták, hogy az NMJ kevésbé volt hatással az érzékenységre, mivel a szekvenálási mélység nőtt. A circode érzékenysége is nagyobb volt, ha nagyobb szekvenálási mélységű Ribo-seq adatokat használtak.
a CircCode összehasonlítása más eszközökkel
a CircCode más eszközökkel, például a CircPro-val való összehasonlításához az A. thaliana Ribo-seq adatainak (SRR3495999) azonos készletét használták a lefordított circrns-ek azonosítására hat processzor segítségével, 16 gigabájt RAM-mal. A CircPro 44 lefordított circrns-t azonosított 13 perc alatt, míg a CircCode 76 lefordított circrns-t azonosított 20 perc alatt. Így a CircCode érzékenyebb, mint a CircPro ugyanazon a számítógépes hardver szinten, de több időt vesz igénybe. A CircPro tömör és kevésbé időigényes, mint a CircCode, de a CircCode több circrns-t képes azonosítani kódolási képességgel, mint a CircPro.
következtetések
a Circrns-ek fontos szerepet játszanak a biológiában, és döntő fontosságú a circrns-ek pontos azonosítása a kódolási képességgel a későbbi kutatásokhoz. Alapján Python 3, kifejlesztettük CircCode, egy könnyen használható parancssori eszköz, amely nagy érzékenységgel azonosítására lefordított circrns származó Ribo-Seq olvas nagy pontossággal. A circode jó teljesítményt mutat mind növényekben, mind állatokban. A jövőbeni munka hozzáadja a downstream karakterelemzést a CircCode-hoz a folyamat minden lépésének megjelenítésével és az előrejelzés pontosságának optimalizálásával.
elérhetőség és követelmények
a CircCode elérhető https://github.com/PSSUN/CircCode címen; operációs rendszer(ek): Linux, programozási nyelvek: Python 3 és R; egyéb követelmények: bedtools (2.20.0 vagy újabb verzió), bowtie, STAR, Python 3 csomagok (Biopython, Pandas, rpy2), R-csomagok (BASiNET, Biostrings). Az összes szükséges szoftver telepítőcsomagja elérhető a CircCode honlapján. A felhasználóknak nem kell külön letölteniük őket. A CircCode kezdőlapja részletes felhasználói kézikönyveket is tartalmaz referenciaként. Az eszköz szabadon elérhető. Nincsenek korlátozások a nonacademics használatára.
adatok rendelkezésre állásáról szóló nyilatkozat
minden vonatkozó adat a kéziratban és az azt alátámasztó információs fájlokban található.
szerzői hozzájárulások
Konceptualizáció: PS, GL. Adatkezelés: PS, GL. Hivatalos elemzés: PS, GL. Írás-eredeti tervezet: PS, GL. Írás-áttekintés és szerkesztés: PS, GL.
finanszírozás
ezt a munkát támogatta támogatások a National Natural Science Foundation of China (grant nos. 31770333, 31370329, és 11631012), a Program New Century kiváló tehetségek egyetemi (NCET-12-0896), és az alapvető kutatási alapok a központi egyetemek (nem. GK201403004). A finanszírozó ügynökségeknek nem volt szerepük a tanulmányban, annak megtervezésében, az adatgyűjtésben és-elemzésben, a közzétételről szóló döntésben vagy a kézirat elkészítésében. A finanszírozóknak nem volt szerepük a tanulmány tervezésében, az adatgyűjtésben és az elemzésben, a közzétételről szóló döntésben vagy a kézirat elkészítésében.
összeférhetetlenség
a szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségnek tekinthetők.
Kiegészítő anyag
a cikk kiegészítő anyaga megtalálható az interneten:: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
kiegészítő adatok 1 / az előre jelzett transzlált circrns és rövid peptid szekvenciája.
kiegészítő adatok 2 | GO dúsítási és KEGG dúsítási eredmények az emberek és az Arabidopsis thaliana esetében.
kiegészítő adatok 3 | a becsült lefordított circrns-ek összehasonlítása validált translated circrns-ekkel.
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: rugalmas trimmer az illumina szekvencia adatokhoz. Bioinformatika 30, 2114-2120. doi: 10.1093 / bioinformatika / btu170
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Brar, G. A., Weissman, J. S. (2015). A riboszóma profilozás feltárja a fehérjeszintézis mit, mikor, hol és hogyan. Nat. Mol Tiszteletes. Sejt Biol. 16, 651–664. doi: 10.1038 / nrm4069
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Chen, L.-L., Yang, L. (2015). A circrns biogenezisének szabályozása. RNS Biol. 12, 381–388. doi: 10.1080/15476286.2015.1020271
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Chu, Q., Zhang, X., Zhu, X., Liu, C., Mao, L., Ye, C., et al. (2017). PlantcircBase: adatbázis a növényi kör alakú RNS-ekhez. Mol. 10-es üzem, 1126-1128. doi: 10.1016 / j. molp.2017.03.003
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Ib, S., et al. (2013). Csillag: ultragyors univerzális RNS-seq aligner. Bioinformatika 29, 15-21. doi: 10.1093 / bioinformatika / bts635
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Dong, R., Ma, X.-K., Chen, L.-L., Yang, L. (2019). “A circrns-ek Genom-szintű annotációja és alternatív visszacsatolásuk / összekapcsolásuk a CIRCexplorer Pipeline-rel”, az Epitranscriptomics-ban. Eds. Wajapeyee, N., Gupta, R. (New York, NY: Springer New York), 137-149. doi: 10.1007/978-1-4939-8808-2_10
CrossRef teljes szöveg / Google Tudós
Dong, R., Ma, X.-K., Li, G.-W., Yang, L. (2018). CIRCpedia v2: frissített adatbázis az átfogó körkörös RNS annotációhoz és expressziós összehasonlításhoz. Genomika Proteomika Bioinf. 16, 226–233. doi: 10.1016 / j.gpb.2018.08.001
CrossRef Teljes Szöveg / Google Scholar
Gao, Y., Wang, J., Zhao, F. (2015). CIRI: hatékony és elfogulatlan algoritmus a De novo körkörös RNS azonosításához. Genome Biol. 16, 4. doi: 10.1186 / s13059-014-0571-3
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Guttman, M., Russell, P., Ingolia, N. T., Weissman, J. S., Lander, E. S. (2013). A riboszóma profilozás bizonyítékot szolgáltat arra, hogy a nagy nem kódoló RNS-ek nem kódolják a fehérjéket. 154-es cella, 240-251. doi: 10.1016 / j. cella.2013.06.009
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Scholar
Hsu, P. Y., Benfey, P. N. (2018). Kicsi, de hatalmas: funkcionális peptidek, amelyeket kis ORF-ek kódolnak a növényekben. Proteomika 18, 1700038. doi: 10.1002 / pmic.201700038
CrossRef teljes szöveg / Google Scholar
Hsu, P. Y., Calviello, L., Wu, H.-Y. L., Li, F. W., Rothfels, C. J., Ohler, U. és mtsai. (2016). A szuperfelbontású riboszóma-profilozás nem jegyzett fordítási eseményeket tár fel az Arabidopsis – ban. Proc. NAT. Acad. Sci. 113, E7126-E7135. doi: 10.1073 / pnas.1614788113
CrossRef Teljes Szöveg / Google Scholar
Ingolia, N. T., Lareau, L. F., Weissman, J. S. (2011). Az egér embrionális őssejtek riboszóma profilozása feltárja az emlősök proteomjainak összetettségét és dinamikáját. 147-es, 789-802-es cella. doi: 10.1016 / j. cella.2011.10.002
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Ito, E. A., Katahira, I., Vicente, F. F., da, R., Pereira, L. F. P., Lopes, F. M. (2018). BASiNET-biológiai szekvenciák hálózata: esettanulmány a kódoló és nem kódoló RNS-ek azonosításáról. Nukleinsavak res. 46, e96-e96. doi: 10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093 / bioinformatika / bty948
CrossRef teljes szöveg / Google Scholar
Kearse, M. G., Wilusz, J. E. (2017). Nem AUG fordítás: a fehérjeszintézis új kezdete az eukariótákban. Gének Dev. 31, 1717–1731. doi: 10.1101 / gad.305250.117
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Csong, L., Csang, Y., Ye, Z.-Q., Liu, X.-Q., Zhao, S.-Q., Wei, L., et al. (2007). CPC: értékelje az átiratok fehérjekódoló potenciálját szekvencia jellemzők és támogató vektor gép segítségével. Nukleinsavak Res. 35, W345-W349. doi: 10.1093 / nar / gkm391
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Langmead, B., Salzberg, S. L. (2012). Gyors gapped-olvassa igazítás Bowtie 2. Nat. 9. módszer, 357-359. doi: 10.1038 / nmeth.1923
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós
Meng, X., Chen, Q., Zhang, P., Chen, M. (2017). CircPro: integrált eszköz a fehérje-kódoló potenciállal rendelkező circrns-ek azonosítására. Bioinformatika 33, 3314-3316. doi: 10.1093 / bioinformatika / btx446
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Michel, A. M., Baranov, P. V. (2013). Riboszóma profilozás: Hi-Def monitor a fehérjeszintézishez a genom egészére kiterjedő skálán: riboszóma profilozás. Wiley Interdiscip. Rev. RNS 4, 473-490. doi: 10.1002 / wrna.1172
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
B. A., Bartók, O., Jens, M., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., et al. (2017). CircRNAs fordítása. Mol. 66-os cella, 9-21.e7. doi: 10.1016 / j.molcel.2017.02.021
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós
Ponty, Y., Termier, M., Denise, A. (2006). GenRGenS: szoftver generáló véletlen genomiális szekvenciák és struktúrák. Bioinformatika 22, 1534-1535. doi: 10.1093 / bioinformatika / btl113
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Rho, M., Tang, H., Ye, Y. (2010). FragGeneScan: a gének előrejelzése rövid és hibára hajlamos olvasmányokban. Nukleinsavak res. 38, e191-e191. doi: 10.1093 / nar / gkq747
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Slavoff, S. A., Mitchell, A. J., Schwaid, A. G., Cabili, M. N., Ma, J., Levin, J. Z., et al. (2013). Rövid nyitott olvasókeret-kódolt peptidek peptidomikus felfedezése az emberi sejtekben. Nat. Kémia. Biol. 9, 59–64. doi: 10.1038 / nchembio.1120
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Schiffer András, A. W., May, G. E., Kuersten, S., Freeberg, L., Murphy, R. F., et al. (2018). Konzervált, nem AUG uorf-ok, amelyeket a riboszóma profilozási ADATOK új regressziós elemzése tárt fel. Genom Res. 28, 214-222. doi: 10.1101 / g.221507.117
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós
Vicens, Q., Westhof, E. (2014). A körkörös RNS-ek biogenezise. 159-es cella, 13-14. doi: 10.1016 / j. cella.2014.09.005
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Willems, P., Ndah, E., Jonckheere, V., Stael, S., Matrica, A., Martens, L., et al. (2017). Az N-terminális proteomika segítette az Arabidopsis thaliana feltáratlan fordítási beavatási tájának profilozását. Mol. Cella. Proteomika 16, 1064-1080. doi: 10.1074 / mcp.M116. 066662
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós
Wu, J., Mao, X., Cai, T., Luo, J., Wei, L. (2006). KOBAS server: web – alapú platform automatizált annotációhoz és útvonal azonosításhoz. Nukleinsavak Res. 34, W720-W724. doi: 10.1093 / nar / gkl167
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
Yang, L., Fu, J., Zhou, Y. (2018). Körkörös RNS-ek és azok kialakuló szerepe az Immunszabályozásban. Elöl. Immunol. 9, 2977. doi: 10.3389 / fimmu.2018.02977
PubMed absztrakt / CrossRef teljes szöveg / Google Scholar
Yang, Y., rajongó, X., Mao, M., dal, X., Wu, P., Zhang, Y., et al. (2017). Az N6-metiladenozin által vezérelt körkörös RNS-ek kiterjedt fordítása. Res. 27-Es, 626-641-Es Cella. doi: 10.1038 / KR.2017.31
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós
Yu, G., Wang, L.-G., Han, Y., Ő, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar