Inleiding
de Limb-Girdle Muscular Dystrophies (Lgmd’ s) omvatten een heterogene groep aandoeningen die gekenmerkt worden door de progressieve verzwakking en verzwakking van de proximale limb-girdles spieren (1, 2). Lgmd ‘ s vertonen inter-en intrafamiliale variabiliteit, variërend van zeer milde vormen tot ernstige, vroeg beginnende, snel progressieve fenotypen (2). Lgmd ‘ s kunnen worden geclassificeerd als autosomaal dominant (LGMD1) of recessief (LGMD2). De eerste groep heeft meestal een volwassen beginleeftijd en komt niet erg vaak voor (<10% van alle Lgmd ‘ s), terwijl de laatste vaker voorkomen (1:15000) (1). Onder recessieve vormen is LGMD2A (of calpainopathie) wereldwijd de meest voorkomende LGMD, die ongeveer 30% van alle lgmd-gevallen treft (3). Klinische tekenen van de ziekte zijn onder meer tiptoe lopen, waggelen gang, moeite met rennen en het beklimmen van trappen, scapulier winging. Gewrichtscontracturen, verkorting van de achillespees, scoliose worden vaak waargenomen, terwijl gezichts-en nekspieren niet worden aangetast (4). Een asymptomatische Hyperckemie (5-80 keer CPK normale niveaus) bij jonge patiënten wordt beschouwd als een preklinisch stadium van de ziekte en kan aanhouden voor meerdere jaren (1, 5). De beginnende leeftijd van spierzwakte treedt gewoonlijk op na 15 jaar, hoewel dit zich kan voordoen op oudere (<12 jaar) of latere (>30 jaar) leeftijd. De ziekteprogressie kan leiden tot verlies van ambulatie, respiratoire insufficiëntie en verminderde vitale capaciteit van de longen in de gevorderde stadia (6). Betrokkenheid van het hart (hartritmestoornissen, hartgeleidingsstoornissen, disfunctie van de linkerventrikel-ejectie) wordt slechts incidenteel gemeld (6, 7).
de diagnose calpainopathie wordt bevestigd door de detectie van pathogene mutaties in CAPN3 (15q15.1) (4), coderend voor verschillende, alternatief gesplitste transcripten. Het volledige transcript wordt echter voornamelijk uitgedrukt in spierweefsel (8). Het gecodeerde eiwit (CAPN3) is een lid van niet-lysosomale CA++-afhankelijke cysteine proteasen familie. In spier, neemt CAPN3 deel aan” sarcomere het remodelleren, ” die voor spieraanpassing en de groei in antwoord op functionele en metabolische eisen essentieel is. Tot op heden zijn meer dan 490 pathogene mutaties in CAPN3 beschreven, waarvan de meeste single-nucleotide veranderingen zijn (4). Mutaties in CAPN3 zijn geassocieerd met mitochondriale afwijkingen, groeifalen, verhoogde oxidatieve stress en sarcomere desorganisatie die helemaal bijdragen aan de spier niet in staat om belastingen vast te houden, waardoor myofiber degeneratie en spierafbraak (8). De diagnose van calpainopathie kan uitdagend zijn wegens de genetische heterogeniteit en de niet-specificiteit van klinisch en instrumenteel patroon. De verdeling van spierzwakte/atrofie, hypertrofie/pseudohypertrofie en peescontracturen wordt zeer vaak gedeeld met andere LGMDs of neuromusculaire aandoeningen. Het patroon van de spierbiopsie in calpainopathie is over het algemeen ook niet-specifiek, variërend van milde spierafwijkingen aan strenge dystrofische veranderingen. Bovendien zijn immunohistochemische / biochemische merkers meestal niet betrouwbaar: het signaal van calpain kan zelfs in aanwezigheid van een niet-functioneel eiwit normaal zijn, en omgekeerd kan het zelfs in andere spierdystrofieën worden verminderd verschillend van calpainopathie.
het wordt daarom aanbevolen een differentiële diagnose uit te voeren om nauwkeurige en betrouwbare resultaten te verkrijgen. Hiertoe zijn moleculaire genetische testbenaderingen ontwikkeld om de diagnose van calpainopathie te bevestigen, met inbegrip van multigeenpaneel, uitgebreide genomische (exoom/genoomsequencing) en single-genanalyse (directe sequencing) (9). De implementatie van Next-Generation Sequencing (NGS) was nuttig om informatieve gegevens te genereren voor diagnostische, voorspellende of therapeutische doeleinden (10-12). De genpanelen van NGS zijn gebaseerd op de analyse van een reeks genen verbonden aan een specifieke ziekte of een groep verwante wanorde, die door genetische en fenotypic heterogeniteit worden gekenmerkt. NGS-panelen kunnen ook nuttig zijn voor het opsporen van gemengde of complexe fenotypen, die het gevolg zijn van de overerving van meer dan één genetisch defect van ouders (13). Over het algemeen komen patiënten met complexe fenotypen, die negatief zijn voor de bekende mutaties in op maat ontworpen diagnostische genpanelen en een brede differentiële diagnose vereisen, in aanmerking voor volledige exoom-of genoomsequencing. Het gehele exoom / genoom rangschikken zijn duurdere benaderingen in termen van gegevensbeheer, interpretatie en analytische kosten maar verhoogt de waarschijnlijkheid van het verstrekken van een moleculaire diagnose aan een vermoede genetische wanorde (13). Wat Lgmd ‘ s betreft, worden speciale NGS-panels ten zeerste aanbevolen om te zorgen voor hoge diagnostische percentages, optimale dekking, gevoeligheid en specificiteit van klinische tests (9). NGS-panels vormen daarom een van de beste systemen om differentiële diagnose te vergemakkelijken, nieuwe causatieve mutaties te identificeren en de correlaties tussen genotype en fenotype te verduidelijken (14, 15).
in dit manuscript wordt het geval van een patiënt met LGMD2A gerapporteerd, waarvoor de toepassing van het NGS-panel niet alleen de diagnose calpainopathie (mutaties in CAPN3) kon bevestigen, maar ook een bijkomende, nieuwe mutatie in het LMNA-gen geassocieerd met gedilateerde cardiomyopathie kon identificeren. Gezien deze resultaten werd de analyse uitgebreid tot de familieleden van de proband om een uitgebreidere interpretatie te geven van de analytische gegevens met betrekking tot de pathologische fenotypen.
Casuspresentatie
klinische karakterisering van Proband en verwanten
het begin van de ziekte bij proband werd verwezen op de leeftijd van 10 jaar, toen zij de aanwezigheid van kalfshypertrofie met tiptoe lopen, moeilijkheden bij lopen, traplopen en opstaan rapporteerde. De symptomen vertoonden een langzame maar progressieve verslechtering, met daaropvolgende betrokkenheid van de proximale bovenste ledematen. Op de leeftijd van 13, werden hoge niveaus van CPK (~8.000 UI/L) toevallig ontdekt, nooit geassocieerd met myoglobinurie. Achtereenvolgens onderging de patiënt verschillende neuromusculaire onderzoeken in verschillende gespecialiseerde centra. Er werden twee spierbiopten uitgevoerd met een klassiek dystrofisch beeld (hypertrofische/atrofische vezels, interne kernen, necrotische vezels, toename van bindweefsel) met niet-specifieke kenmerken. Zoals verwacht waren onderzoeken naar immunochemie en immunoblot niet overtuigend, wat wijst op abnormaal / verminderd α, β, γ sarcoglycaan, β-dystroglycaan en dystrofine signaal alleen in necrotische vezels (herhaalde immunochemie resulteerde normaal, ook voor laminine).; immunobloten voor dystrofine en calpain onthulden normale signalen. Nochtans, is het calpain-3 immunoblot gekend om niet-volledig gevoelig voor lgmd2a diagnose te zijn, aangezien 20-30% van gevallen een normale hoeveelheid proteã ne (1, 16-18) tonen. Vanwege de klinische presentatie (hyperckmie, kuithypertrofie/pseudohypertrofie) werden eerst dystrofinopathieën uitgesloten. Bovendien toonde analyse van DMD (Xp21), FKRP (19q13.32) en DYSF (2p13.2) genen geen pathogene mutaties aan. Patiënt kwam tot onze observatie op de leeftijd van 30, presenteren axiale en gordel betrokkenheid zowel in de bovenste en onderste ledematen, met aanzienlijke waggelende gang en gevleugelde scapulae. In de onderste ledematen was zwakte prominent aanwezig in de quadriceps-en bilspieren die significant IPO-atrofisch waren, samen met het bewijs van “blijkbaar hypertrofische” (pseudohypertrofie) kuitspieren (figuur 1). Krampen, myalgia ‘ s en rimpelvorming werden niet waargenomen. Volledige pneumologische (spirometrie en polysomnografie) en cardiologische (echografie, 24-uur ECG Holter monitoring, cardiale MRI, volledige cardiologische fysieke inspectie met gerichte medische anamnese en cardiovasculaire reflex analyse) onderzoeken hebben geen significante afwijkingen aan het licht gebracht. We hebben ook zuurmaltase deficiëntie uitgesloten (normale enzymactiviteit assays in lymfocyten/leukocyten). Muscle Magnetic Resonance Imaging (MRI) toonde een opvallende betrokkenheid van scapulaire gordel, paravertebrale spieren, achterste compartiment spieren van de dij (met relatieve sparing van de sartorius en gracilis spieren) en van het been (in het bijzonder gastrocnemius medialis en soleus) (Figuur 2). Dit patroon van betrokkenheid is al gemeld in de literatuur over MRI-foto ‘ s bij calpainopathie (19, 20). Bovendien toonde de MRI-studie aan dat de kuitspieren effectief atrofisch waren en gekenmerkt werden door vetinfiltratie, wat een spieruitbreiding veroorzaakte.”De klinische presentatie, de aanvangsleeftijd, de snelheid van progressie, de verdeling van spierzwakte en de MRI-bevindingen in de proband waren volledig consistent met een diagnose van LGMD2A die uitgebreid beschreven werd in de Literatuur (4, 21, 22).
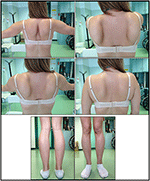
figuur 1. Proband klinische kenmerken: gevleugelde scapulae en combinatie van dij atrofie en kuit pseudo-hypertrofie.
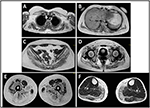
Figuur 2. Proband spier MRI: betrokkenheid van de gordel van de bovenste ledematen en de paravertebrale spieren (A,B), betrokkenheid van de gordel van de onderste ledematen en het achterste compartiment van de dij (C–E, meestal de glutei) met relatief sparen van de sartorius en gracilis, en betrokkenheid van het achterste compartiment van het been (F, meestal gastrocnemius medialis/soleus).
uit de evaluatie van de familieleden van de proband bleek geen voorgeschiedenis van neuromusculaire ziekte en geen bewijs van bloedverwantschap tussen ouders. Echter, de moeder van de proband, op de leeftijd van 55, presenteerde 2 syncopale episodes waarvoor ze werd gediagnosticeerd met idiopathische bradycardie (tot 40 bpm ‘ s nachts). Uitgebreide cardiologische onderzoeken op de moeder van de proband gediagnosticeerd een symptomatische atrioventriculaire (AV) blok na het optreden van verschillende syncopale episodes/lipothymie. De analyse toonde de aanwezigheid aan van een basis eerstegraads AV-blok, met verscheidene perioden van ernstige bradycardie, meestal nachtelijk en vaak symptomatisch, geassocieerd met enkele fasen van tweedegraads AV-blok, zowel type 1 als 2, en enkele fasen van nachtelijke volledige av-dissociatie. Echocardiografie en ergometrische test toonden geen significante veranderingen, behalve voor het patent foramen ovale. De moeder vertoonde geen andere klinische symptomen, predisponerende toestand of risicofactor. Gezien haar ziektebeeld, werd de moeder van de proband geïmplanteerd met PMK. Bovendien, familiegeschiedenis bleek dat de grootmoeder en de overgrootvader van de proband werden ook geïmplanteerd met PMK op de leeftijd van 55 en 30 jaar, respectievelijk. Bovendien stierf de broer van de grootmoeder van de proband op 51-jarige leeftijd voor ernstige gedilateerde cardiomyopathie. Cardiologische onderzoeken werden ook uitgevoerd op de vader en de oom van de moeder van de proband die volledig onaangetast resulteerde.
deze studie werd goedgekeurd door de ethische commissie van de Santa Lucia Foundation en werd uitgevoerd volgens de Verklaring van Helsinki. Alle deelnemers verstrekten ondertekende geïnformeerde toestemming voor genetische analyse, en in dit verband verstrekten zij ook toestemming voor publicatie van dit gevalsrapport.
laboratoriumonderzoeken en diagnostische Tests
genomisch DNA werd geëxtraheerd uit perifeer bloed (400 µL) met behulp van MagPurix Bloeddna-Extractiekit en MagPurix automatisch extractiesysteem (Resnova) volgens de instructies van de fabrikant. De steekproeven werden gerangschikt gebruikend Ion PGM systeem en Ion Ampliseq aangepast paneel hoge specificiteit (Thermo Fisher Scientific). De grootte van het paneel was 129.13 Kb, die naar verwachting ~99,72% van het totale panel met een minimale dekking van 20X scherm. het panel omvatte 18 genen, die werden geselecteerd met behulp van wetenschappelijke literatuur, GeneReviews (www.ncbi.nlm.nih.gov/books/NBK1116/) en frequentie van pathogene varianten in de algemene bevolking. Een gedetailleerde beschrijving van het NGS-panel is samengevat in Tabel 1.
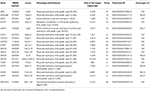
Tabel 1. Aangepast NGS-paneel gebruikt voor lgmd-diagnose.
bibliotheken werden gebouwd door Ion AmpliSeq™ Library Kits 2.0. Ongeveer 10 ng / µl van beginnend DNA werden gebruikt voor multiplex PCR reacties. Achtereenvolgens, werden twee zuiveringsstappen (gebruikend AMPure XP, Beckman Coulter) uitgevoerd om ongewenste verontreinigende stoffen te verwijderen en een definitieve PCR werd uitgevoerd. Template versterking en verrijking stappen werden uitgevoerd door Ion PGM Hi-Q OT2 kit-400, Ion OneTouch 2 systeem en Ion OneTouch ES (Thermo Fisher Scientific). De steekproeven werden verwerkt door Ion PGM Hi-Q Het rangschikken uitrusting (400 bp, Thermo Fisher Scientific) en lopen op Ion 316 Spaander v2 (vereiste 850 stromen) en Ion PGM Sequencer (Thermo Fisher Scientific). De resultaten werden geanalyseerd gebruikend Ionenverslaggever 4.6 (Thermo Fisher Scientific) en geïntegreerde Genoomviewer (IGV). De interpretatie van genetische varianten werd uitgevoerd door Human Gen Mutation Database (Hgmd), Leiden Open Variation Database (LOVD), ClinVar en ExAC. Het functionele effect van de gedetecteerde varianten werd geëvalueerd door bioinformatische voorspellende hulpmiddelen, met inbegrip van Mutatieproever, Varsome, zift, Polyfen 2, SMART, Human Splicing Finder (HSF). Directe sequencing (BigDye Terminator v3.1, BigDyeX Terminator en ABI3130, Applied Biosystems) werd uitgevoerd om genetische varianten te bevestigen en om genomische codeergebieden met een dekking <20X te sequencen (LMNA, Chr1:156106052-156106076; DYSF, Chr2:71753352-71753502, Chr2:71776385-71776640; SGCB, Chr4:52904225-52904560; SGCA, Chr17:48243242-48243570).
resultaten
de NGS-analyse van de proband bracht 3 heterozygote varianten aan het licht (aanvullend figuur 1). Twee varianten werden gelokaliseerd in CAPN3, namelijk NM_000070. 2 (CAPN3): C.550delA (p.Thr184Argfs) en C.1813G>C (P.Val605Leu) in respectievelijk de exons 4 en 16. De eerste is een enkele nucleotide deletie en is de meest voorkomende (75% van de gevallen) pathogene variant in Europese landen (1). Zoals verwacht classificeerde de bioinformatische analyse de C.550delA (rs80338800) als een loss-of-function variant (p.Thr184Argfs), waardoor een frameverschuiving van het Open leesframe ontstond. De voorspellende tools (Mutatieproever, HSF, Varsome, Polyfen 2, SIFT) beschreven de variant als schadelijk. Bovendien onthulde SMART dat het veranderde eiwitproduct het calpain 3-domein en de drie “EF-handen” – motieven mist. Het eerste domein is betrokken bij de signalerende wegen van Calpain terwijl de EF-handen essentieel zijn voor de CA++-afhankelijke activering van de proteã ne (4).
met betrekking tot c.1813G>C, is het een nieuwe missense variant (p.Val605Leu) waarvan is voorspeld dat het schadelijk is (door Mutatieproever, HSF, Varsome, Polyfen 2, SIFT) vanwege een mogelijke wijziging van splicing. Deze variant is niet geannoteerd in de literatuur of in online databases en is niet gevonden bij 200 proefpersonen in de controlegroep. Helaas leverde de analyse door SMART tool geen significante resultaten op, omdat de variant zich binnen een niet-gekarakteriseerd domein bevindt. De segregatie-analyse van CAPN3 toonde aan dat de moeder en de oom van de moeder beiden heterozygoot waren voor ca.550delA, terwijl de vader drager was van ca.1813G>C.
bovendien bleek uit NGS-analyse van de proband dat er een nieuwe variant in LMNA aanwezig was, namelijk NM_170707 (LMNA): c.550C>T (p.Gln184*). De te veel vermelde variant is voorspeld als een nulvariant, is nog niet beschreven in de literatuur of in online databases en is niet gevonden bij 200 proefpersonen in de controlegroep. Voorspellende tools (Mutatieproever, HSF, Varsome, Polyfen 2, zift) beschreven een pathogeen effect van deze variant op het eiwitproduct. Het slimme hulpmiddel meldde dat de veranderde LMNA-proteã ne het gloeidraaddomein mist, dat essentieel is om zijn structuur en functie te handhaven. De segregatieanalyse wees op de aanwezigheid van de c.550C>t variant alleen bij de moeder, wat wijst op een mogelijk verband met haar cardiale symptomatologie en haar familiegeschiedenis van de ziekte.
volgens de criteria vastgesteld door de American College of Medical Genetics (ACMG) Standards and Guidelines (23), c. 1813G>C (CAPN3) kan worden beschreven als een waarschijnlijke pathogene variant, gezien het feit dat het zich bevindt in een kritisch domein zonder goedaardige variatie (PM1); het is afwezig in ExAc, GnomAD en 1000 Genome Browser databases (PM2); gezien recessief model van overerving, is het ontdekt in trans met een pathogene variant (PM3); meerdere lijnen van computationele bewijs ondersteunen een schadelijk effect op het gen of genproduct (PP3). Wat de klinische classificatie van C.550C>T (LMNA) door ACMG betreft, kan het worden aangeduid als een pathogene variant, aangezien het een nulvariant is die functieverlies veroorzaakt (p.Gln184*) in LMNA (PVS1); het is afwezig in ExAc, GnomAD en 1000 Genome Browser databases (PM2); het bevindt zich in een kritisch domein zonder goedaardige variatie (PM1); meerdere regels van computationeel bewijs ondersteunen een schadelijk effect op het gen of genproduct (PP3).
conclusies
in dit casusrapport werd een patiënt behandeld met LGMD, die drager bleek te zijn van mutaties, niet alleen in CAPN3 (c.550delA en c.1813G>C), maar ook in LMNA (c.550C>T). Deze resultaten verklaren het neuromusculaire fenotype van de proband vanwege CAPN3 mutaties en wijzen op een potentieel risico op cardiovasculaire aandoeningen als gevolg van de aanwezigheid van de variant in LMNA en haar positieve familiegeschiedenis. Gezien deze resultaten werden de familieleden onderworpen aan de segregatieanalyse. In het bijzonder bleek de moeder drager te zijn van respectievelijk CAPN3_c.550delA en LMNA_c.550C>T. De moeder kan worden beschouwd als een gezonde drager voor de capn3_c.550delA pathogene mutatie geassocieerd met calpainopathie. Echter, de aanwezigheid van een nieuwe variant in LMNA kan verklaren haar cardiovasculaire pathologie (bradycardie en syncopale episodes). De afwezigheid van neuromusculaire symptomatologie bij de moeder en het eigenaardige klinische beeld van de proband sluit een mogelijke associatie van LMNA_c uit.550C> T met neuromusculair fenotype, in het bijzonder met betrekking tot musculaire dystrofie van Emery-Dreifuss (EMD). In feite beïnvloedt EMD specifiek vastus lateralis en biceps brachii spieren die relatief gespaard worden bij LGM2A (24, 25).
de oom van moederszijde was alleen heterozygoot voor de CAPN3_c.550delA en vertoonde, zoals verwacht, geen neuromusculaire of cardiovasculaire problemen. De vader kreeg de roman CAPN3_c.1813G>C en daardoor werd hij niet beïnvloed. Over het geheel genomen bevestigde segregatieanalyse de overerving van de drie mutaties van de proband van haar familieleden en wees op een vertrouwdheid voor cardiomyopathie die niet kan worden verwaarloosd (Figuur 3).
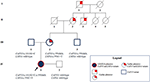
Figuur 3. Stamboom die de positieve vertrouwdheid toont voor cardiaal fenotype geërfd door maternale afstamming en de overdracht van de CAPN3-en LMNA-varianten door de familieleden.
in totaal doen deze gegevens enkele belangrijke overwegingen rijzen. Ten eerste was het NGS-panel in deze patiënt van cruciaal belang om de moleculaire diagnose van calpainopathie te bereiken, waarbij een bekende mutatie en een tweede nieuwe variant als pathogeen worden voorspeld. Ten tweede, NGS panel toegestaan de identificatie van de nieuwe lmna_c. 550C>T variant in de proband. Dit resultaat was consistent met de positieve familiegeschiedenis en met segregatiegegevens, wat hart-en vaatziekten bij de moeder verklaart en, nog belangrijker, meer specifieke cardiologische follow-up bij de proband aanbeveelt. Het is belangrijk op te merken dat cardiologische manifestaties plaatsvonden bij de moeder later in de leeftijd (55 jaar), zodat we kunnen veronderstellen dat de proband nog steeds in een “asymptomatische cardiologische fase zou kunnen zijn.”Echter, een variabele fenotypische expressie van LMNA mutatie in de proband in vergelijking met de moeder kan niet worden uitgesloten. Al deze gegevens onderstrepen het belang van een geïntegreerde aanpak tussen clinici en genetici, voor een juiste interpretatie van de resultaten, een goede genetische counseling en, uiteindelijk, klinische behandeling en follow-up. Met betrekking tot genetische en familiale counseling werd de NGS-analyse ook uitgevoerd op de partner van de proband, om het reproductieve risico voor het paar in te schatten. De partner was negatief voor alle 18 geteste genen, wat betekent dat hij 1/650 restrisico heeft om een gezonde drager te zijn voor lgmd-causatieve mutaties, aangezien de test 84% gevoelig is. Rekening houdend met het genetische profiel van de proband en de gevoeligheid van de NGS-test, is het resterende risico voor het paar om een kind met calpainopathie te krijgen 1/1300. Anderzijds worden de pathogene varianten van LMNA overgedragen volgens een autosomaal dominant patroon, wat betekent dat de proband 50% kans heeft om heterozygote kinderen te hebben. Het klinische beeld van de nakomelingen kan echter niet met zekerheid worden voorspeld aangezien de lmna_c.550C>T een nieuwe variant is en de functionele impact ervan op het fenotype nog onbekend is.
concluderend wordt in dit casusrapport gewezen op het klinische nut van NGS-panels om nauwkeurige lgmd2a-diagnose te stellen en complexe fenotypen en comorbiditeiten te beschrijven die voortkomen uit de overerving van verschillende mutaties in meerdere genen. De toepassing van NGS in de klinische praktijk dient echter altijd te worden gecombineerd met een pre – en post-genetische counseling om een duidelijke uitleg te geven van de resultaten, de mogelijke implicaties op het fenotype van patiënten, het risico op recidief binnen de familie en om mogelijke onverwachte bevindingen te verklaren.
beschikbaarheid van gegevens
voor deze studie werden geen datasets gegenereerd of geanalyseerd.
ethische verklaring
deze studie werd uitgevoerd in overeenstemming met de aanbevelingen van de ethische commissie van de Santa Lucia Foundation met schriftelijke geïnformeerde toestemming van alle proefpersonen. Alle proefpersonen gaven schriftelijke geïnformeerde toestemming overeenkomstig de Verklaring van Helsinki. Het protocol werd goedgekeurd door de ethische commissie van de Santa Lucia Foundation.
Auteursbijdragen
RC, CSt, VC, GC, RG, GPa, SC, CP en JM leverden bijdragen aan de verwerving van gegevens, analyse en interpretatie van gegevens. CSt, RC, GC, RG, GM en EG zijn betrokken geweest bij het opstellen van het manuscript. SZ, GPr, CSa en SS zijn betrokken geweest bij het verzamelen van klinische gegevens. RC, CSt, VC, GC, RG, GPa, SC, CP, JM, SZ, GPr, GM, CSa, SS, en EG hebben de definitieve goedkeuring van de te publiceren versie gegeven.
financiering
dit werk wordt ondersteund door 5X 2016 National Health Ministry 2.
verklaring inzake belangenconflicten
de auteurs verklaren dat het onderzoek werd uitgevoerd zonder enige commerciële of financiële relatie die als een potentieel belangenconflict kon worden opgevat.
aanvullend materiaal
het aanvullende materiaal voor dit artikel is online te vinden op: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. proteïne en genetische diagnose van limb girdle spierdystrofie type 2A: de opbrengst en de valkuilen. Spierzenuw. (2015) 52:163–73. doi: 10.1002/mus.24682
PubMed Abstract / CrossRef Full Text / Google Scholar
2. Nigro V, Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol. (2014) 33:1–12.
PubMed Abstract / Google Scholar
3. Richard I, Hogrel JY, Stockholm D, Payan CAM, Fougerousse F, Eymard B, et al. Natuurlijke voorgeschiedenis van LGMD2A voor het bepalen van uitkomstmetingen in klinische studies. Ann Clin Transl Neurol. (2016) 3:248–65. doi: 10.1002 / acn3.287
PubMed Abstract / CrossRef Full Text / Google Scholar
4. Angelini C, Fanin M. Calpainopathy. In: Adam MP, Ardinger HH, Pagon RA editors. Algemene informatie. Seattle, WA: University Of Washington (2017) 1-30.
PubMed Abstract / Google Scholar
5. Kyriakides T, Angelini C, Schaefer J, Sacconi S, Siciliano G, Vilchez JJ, et al. EFNS-richtlijnen voor de diagnostische benadering van pauci-of asymptomatische hyperckemie. Eur J Neurol. (2010) 17:767–73. doi: 10.1111 / j. 1468-1331. 2010. 03012.x
CrossRef Full Text / Google Scholar
6. Mori-Yoshimura M, Segawa K, Minami N, Oya Y, Komaki H, Nonaka I, et al. Cardiopulmonale disfunctie bij patiënten met limb-girdle musculaire dystrofie 2A. (2017) 55:465–9. doi: 10.1002/mus.25369
CrossRef Full Text / Google Scholar
7. Okere a, Reddy SS, Gupta S, Shinnar M. A cardiomyopathie bij een patiënt met limb girdle muscular dystrofie type 2A. Circ Heart Failure. (2013) 6: e12-13. doi: 10.1161 / CIRCHEARTFAILURE.112.971424
CrossRef Full Text / Google Scholar
8. Kramerova I, Ermolova N, Eskin A, Hevener A, Quehenberger O, Armando AM, et al. Falen om de transcriptie van genen die nodig zijn voor spieraanpassing op te heffen, ligt ten grondslag aan limb girdle musculaire dystrofie 2A (calpainopathie). Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093 / hmg / ddw086
PubMed Abstract / CrossRef Full Text / Google Scholar
9. Thompson R, Straub V. Limb girdle muscular dystrophies-internationale samenwerkingen voor translationeel onderzoek. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038 / nrneurol.2016.35
PubMed Abstract / CrossRef Full Text / Google Scholar
10. Strafella C, Caputo V, Galota MR, Zampatti S, Marella G, Mauriello S, et al. Toepassing van precisiegeneeskunde bij neurodegeneratieve ziekten. Front Neurol. (2018) 9:701. doi: 10.3389 / fneur.2018.00701
PubMed Abstract / CrossRef Full Text / Google Scholar
11. Cascella R, Strafella C, Caputo V, Errichiello V, Zampatti S, Milano F, et al. Naar de toepassing van precisiegeneeskunde bij leeftijdsgebonden maculadegeneratie. Prog Retin Eye Res. (2017) 63:132-46. doi: 10.1016 / j. preteyeres.2017.11.004
PubMed Abstract / CrossRef Full Text / Google Scholar
12. Cascella R, Strafella C, Longo G, Manzo L, Ragazzo M, De Felici C, et al. Het beoordelen van het individuele risico op LMD met genetische counseling, familiegeschiedenis en genetische testen. Oog. (2017) 32:446–50. doi: 10.1038 / eye.2017.192
PubMed Abstract / CrossRef Full Text / Google Scholar
13. Adams DR, Eng CM. Volgende generatie sequencing om vermoedelijke genetische aandoeningen te diagnosticeren. N Engl J Med. (2019) 379:1353–62. doi: 10.1056 / NEJMc1814955
PubMed Abstract / CrossRef Full Text / Google Scholar
14. Angelini C. neuromusculaire ziekte. Diagnose en ontdekking in limb-girdle spierdystrofie. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038 / nrneurol.2015.230
CrossRef Full Text / Google Scholar
15. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett a, Reddel SW, et al. Gebruik van whole-exome sequencing voor diagnose van limb-girdle spierdystrofie: uitkomsten en geleerde lessen. JAMA Neurol. (2015) 72:1424–32. doi: 10.1001 / jamaneurol.2015.2274
PubMed Abstract / CrossRef Full Text / Google Scholar
16. Milic a, Daniele N, Lochmüller H, Mora M, Comi GP, Moggio M, et al. Een derde van de LGMD2A biopten heeft een normale calpain 3 proteolytische activiteit zoals bepaald door een in vitro test. Neuromusc Disord. (2007) 17:148–56. doi: 10.1016/j.nmd.2006.11.001
PubMed Abstract / CrossRef Full Text / Google Scholar
17. Lanzillo R, Aurino S, Fanin M, Aguennoz M, Vitale F, Fiorillo C, et al. Vroeg-beginnende calpainopathie met normaal niet-functioneel calpain 3 niveau. Dev Med Child Neurol. (2006) 48:304–6. doi: 10.1017 / S001216220600065X
PubMed Abstract / CrossRef Full Text / Google Scholar
18. Talim B, Ognibene A, Mattioli E, Richard I, Anderson LV, Merlini L. Normal calpain expression in genetically confirmed limb-girdle muscular dystrophy type 2A.Neurology. (2001) 56:692–3. doi: 10.1212 / wnl.56.5.692-a
CrossRef Full Text / Google Scholar
19. Díaz-Manera J, Llauger J, Gallardo E, Illa I. Muscle MRI in muscular dystrophies. Acta Myol. (2015) 34:95–108.
PubMed Abstract / Google Scholar
20. Mercuri E, Bushby K, Ricci E, Birchall DR, Pane M, Kinali M, et al. Spier-MRI-bevindingen bij patiënten met limb girdle musculaire dystrofie met calpain 3-deficiëntie (LGMD2A) en vroege contracturen. Neuromusc Disord. (2005) 15:164–71. doi: 10.1016/j.nmd.2004.10.008
PubMed Abstract / CrossRef Full Text / Google Scholar
21. Magri F, Nigro V, Angelini C, Mongini T, Mora M, Moroni I, et al. De Italiaanse limb girdle spierdystrofie register: relatieve frequentie, klinische kenmerken en differentiële diagnose. Spierzenuw. (2017) 55:55–68. doi: 10.1002/mus.25192
PubMed Abstract / CrossRef Full Text / Google Scholar
22. Chae J, Minami N, Jin Y, Nakagawa M, Murayama K, Igarashi F, et al. Calpain 3 genmutaties: genetische en klinisch-pathologische bevindingen in limb-girdle spierdystrofie. Neuromusc Disord. (2001) 11:547–55. doi: 10.1016/S0960-8966(01)00197-3
PubMed Abstract / CrossRef Full Text / Google Scholar
23. Richards S, Aziz N, Bale S, Bick D, Das s, Gastier-Foster J, et al. Standaarden en richtlijnen voor de interpretatie van sequentievarianten: een gezamenlijke consensusaanbeveling van het American College of Medical Genetics and Genomics en de Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038 / gim.2015.30
PubMed Abstract / CrossRef Full Text / Google Scholar
24. Bonne G, Mercuri E, Muchir A, Urtizberea A, Bécane HM, Recan D, et al. Klinisch en moleculair genetisch spectrum van autosomaal dominante Emery-Dreifuss spierdystrofie als gevolg van mutaties van het Lamine A/C gen. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170::steun-ANA6>3.0.CO; 2-J
PubMed Abstract / CrossRef Full Text / Google Scholar
25. Hong JS, Ki CS, Kim JW, Suh YL, Kim JS, Baek KK, et al. Cardiale dysritmieën, cardiomyopathie en spierdystrofie bij patiënten met Emery-Dreifuss spierdystrofie en limb-girdle spierdystrofie type 1B. J Koreaanse Med Sci. (2005) 20:283–90. doi: 10.3346 / jkms.2005.20.2.283
CrossRef Full Text / Google Scholar