- Introduction
- Methods
- Filtering van ribosomale Profileringsgegevens
- het samenstellen van virtuele genomen
- bepaling van het Ribo-seq Read-Mapped gebied op een junctie (RMRJ) van circRNAs
- opleiding van het Model en de classificatie van Rmrj ‘s
- voorspelling van vertaalde peptiden door Rmrj ‘ s
- resultaten en discussie
- vertaalde circRNAs bij mensen en A. thaliana
- functionele verrijking van mens en A. thaliana circRNAs met Coderingspotentieel
- nauwkeurigheidstest voor CircCode
- invloed van de Ribo-seq Data Sequencing Depth
- vergelijking van CircCode met andere Tools
- conclusies
- beschikbaarheid en vereisten
- Gegevensbeschikbaarheidsverklaring
- Auteursbijdragen
- financiering
- belangenconflict
- aanvullend materiaal
Introduction
Circrnas (circRNAs) zijn een speciaal type noncoding RNA molecule dat een hot research topic is geworden op het gebied van RNA en veel aandacht krijgt (Chen And Yang, 2015). Vergeleken met traditionele lineaire RNAs (die 5′ en 3′ einden bevatten), hebben circRNA molecules gewoonlijk een gesloten cirkelstructuur; die hen stabieler en minder naar voren gebogen aan degradatie maken (Vicens en Westhof, 2014). Hoewel het bestaan van circRNAs enige tijd gekend is, werden deze molecules beschouwd om een bijproduct van het verbinden van RNA te zijn. Nochtans, met de ontwikkeling van hoog-productie het rangschikken en bioinformaticatechnologieën, circRNAs zijn wijd erkend in dieren en installaties (Chen en Yang, 2015) geworden. Recente studies hebben ook aangetoond dat een groot aantal circRNAs kan worden vertaald in kleine peptiden in cellen (Pamudurti et al., 2017) en hebben een sleutelrol ondanks hun soms lage expressieniveau (Hsu and Benfey, 2018; Yang et al., 2018). Hoewel een toenemend aantal circRNAs worden geà dentificeerd, blijven hun functies in planten en dieren over het algemeen worden bestudeerd. Naast hun functies als Mirna lokvogels, circRNAs hebben belangrijk vertalend potentieel, maar geen hulpmiddelen zijn beschikbaar voor specifiek het voorspellen van de Vertalende mogelijkheden van deze molecules (Jakobi en Dieterich, 2019).
er bestaan verschillende instrumenten voor de voorspelling en identificatie van circRNAs, zoals CIRI (Gao et al., 2015), CIRCexplorer (Dong et al., 2019), CircPro (Meng et al., 2017), en circtools (Jakobi et al., 2018). Onder hen, CircPro kan vertaalde circRNAs onthullen door het berekenen van een vertaling potentiële score voor circRNAs op basis van CPC (Kong et al., 2007), dat is een hulpmiddel voor het identificeren van de open reading frame (ORF) in een bepaalde volgorde. Echter, omdat sommige circRNAs het startcodon niet gebruiken tijdens de vertaling (Ingolia et al., 2011; Slavoff et al., 2013; Kearse and Wilusz, 2017; Spealman et al., 2018), het gebruik van CPC kan filteren uit een aantal echt vertaald circRNAs. In deze studie gebruikten we BASiNET (Ito et al., 2018), die een RNA-classifier is die op de machine het leren methodes wordt gebaseerd (random forest en J48 model). Het transformeert aanvankelijk gegeven codage RNAs (positieve gegevens) en noncoding RNAs (negatieve gegevens) en vertegenwoordigt hen als complexe netwerken; het haalt dan de topologische maatregelen van deze netwerken uit en construeert een eigenschap vector om het model te trainen dat wordt gebruikt om de codagecapaciteit van circRNAs te classificeren. Met deze methode, wordt het onjuiste filteren van vertaalde circRNAs vermeden die niet door AUG worden geïnitieerd. Bovendien, Ribo-seq technologie, die op hoog-productie het rangschikken is gebaseerd om rpfs (ribosomal beschermde fragmenten) van afschriften te controleren (Guttman et al., 2013; Brar and Weissman, 2015), kan worden gebruikt om de locaties van circRNAs te bepalen die worden vertaald (Michel en Baranov, 2013). Om het codeervermogen van circRNAs te identificeren, ontwikkelden we de tool CircCode, die een Python 3–gebaseerd kader omvat, en toegepast CircCode om het vertaalpotentieel van circRNAs van mensen en Arabidopsis thaliana te onderzoeken. Ons werk verstrekt een rijke bron voor verdere studie van de functies van circRNAs met codagecapaciteit.
Methods
CircCode is geschreven in de programmeertaal Python 3; Het maakt gebruik van Trimmomatic (Bolger et al., 2014), bowtie (Langmead and Salzberg, 2012), en STAR (Dobin et al., 2013) om ruwe Ribo-seq te filteren leest en deze gefilterde leest aan het genoom in kaart te brengen. CircCode identificeert dan Ribo-seq read-mapped regio ‘ s in circRNAs die juncties bevatten. Na dat, wordt de kandidaat in kaart gebracht opeenvolgingen in circRNAs gesorteerd gebaseerd op classifiers (J48 model) in codage RNAs en noncoding RNAs door BASiNET. Tot slot, worden de korte peptides geproduceerd door vertaling geà dentificeerd als potentiële codagegebieden van circRNAs. Het hele proces van CircCode bestaat uit vijf stappen (figuur 1).
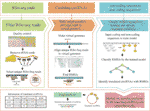
figuur 1 de workflow van CircCode. De bovenste laag vertegenwoordigt het invoerbestand dat nodig is voor elke stap van CircCode. De middelste laag is verdeeld in drie delen, en elk deel vertegenwoordigt een verschillende fase van de operatie. Van links naar rechts vertegenwoordigt het eerste deel de filtering van de Ribo-seq gegevens; de kwaliteitscontrole wordt uitgevoerd door Trimmomatic, en de rRNA reads worden verwijderd door bowtie. Het tweede deel vertegenwoordigt de stappen die worden gebruikt om het virtuele genoom te produceren en gefilterde leest aan het virtuele genoom met ster af te stemmen. Het laatste deel vertegenwoordigt de identificatie van vertaalde circRNAs door machine het leren. De onderste laag vertegenwoordigt de laatste stap die wordt gebruikt om peptides te voorspellen die van circRNAs en de definitieve outputresultaten worden vertaald, met inbegrip van informatie over vertaalde circRNAs en hun vertaal producten.
Filtering van ribosomale Profileringsgegevens
eerst worden fragmenten en adapters van lage kwaliteit in de Ribo-Seq-reads verwijderd door Trimmomatic met de standaardparameters om schone Ribo-seq-reads te verkrijgen. Ten tweede, worden deze schone Ribo-seq reads in kaart gebracht aan een rRNA-bibliotheek om reads te verwijderen die uit rRNA worden afgeleid gebruikend bowtie. Omdat de leeslengtes van Ribo-seq relatief kort zijn (over het algemeen minder dan 50 bp), is het mogelijk voor één gelezen om veelvoudige gebieden aan te passen. In dit geval is het moeilijk om te bepalen met welke regio een bepaalde read overeenkomt. Om dit te vermijden, worden de schone Ribo-seq leest in kaart gebracht aan het genoom van een species van belang, en leest die niet perfect aan het genoom worden afgestemd worden beschouwd als definitieve unieke Ribo-seq leest.
het samenstellen van virtuele genomen
Circrna ‘ s verschijnen gewoonlijk als ringvormige moleculen in eukaryoten, en ze kunnen worden geïdentificeerd op basis van hun back-splicing junctions. Nochtans, zijn de opeenvolgingen van circRNAs in het fasta dossier vaak in lineaire vorm. In theorie, wijst het resultaat erop dat de verbinding tussen eindnucotide 5′ en eindnucotide 3’ is, hoewel de verbinding en de opeenvolging dichtbij de verbinding niet direct kunnen worden bekeken, waarbij Ribo-seq aan circRNA opeenvolgingen, met inbegrip van verbindingen, op een duidelijke manier wordt afgestemd.
CircCode verbindt de sequentie van elke circRNA in tandem zodanig dat de junctie voor elke in het midden van de nieuw geconstrueerde sequentie ligt. Wij scheidden ook elke reekseenheid door 100 N nucleotiden om verwarring bij de opeenvolgingsrichtingsstap te vermijden (de lengte van elke RPF is minder dan 50 bp). Tenslotte verkregen we een virtueel genoom dat alleen bestaat uit kandidaat circRNAs in tandem gescheiden door 100 Ns. Omdat CircCode slechts op aanpassing tussen Ribo-seq leest en circRNA opeenvolgingen concentreert, kunnen wij het codagepotentieel van circRNAs onderzoeken door Ribo-seq leest aan dit virtuele genoom in kaart te brengen, dat een grote hoeveelheid computationele tijd kan besparen (het virtuele genoom is veel kleiner dan het gehele genoom) en de nauwkeurigheid kan verhogen (door interferentie tussen upstream en downstream opeenvolgingsvergelijkingen van circRNAs te vermijden).
bepaling van het Ribo-seq Read-Mapped gebied op een junctie (RMRJ) van circRNAs
de uiteindelijke unieke Ribo-seq reads worden in kaart gebracht op een eerder gecreëerd virtueel genoom met behulp van STAR. Omdat elke tandem circRNA eenheid werd gescheiden door 100 N basissen alvorens het virtuele genoom te produceren,werd de grootste Intron lengte ingesteld om niet meer dan 10 basissen met de parameter “–alignIntronMax 10.”Deze parameter elimineert om het even welke interactie tussen verschillende circRNAs in de opeenvolgingsuitlijning. In de tweede stap van virtuele genoomproductie, slaat CircCode positionele verbindingsinformatie voor elke circRNA in het virtuele genoom op. Als het Ribo-seq read-mapped gebied in het virtuele genoom de verbinding van circRNA omvat, en het aantal in kaart gebrachte Ribo-seq leest op Verbinding (NMJ) groter is dan 3, Kan het Ribo-seq reads-mapped gebied op Verbinding van circRNAs als RMRJ worden beschouwd, die een ruwweg vertaald segment van circRNAs dichtbij de junctieplaats openbaart.
opleiding van het Model en de classificatie van Rmrj ‘s
hoewel Rmrj’ s een krachtig bewijs van Vertaling kunnen vormen, zijn er nog enkele tekortkomingen in deze methode. Omdat de lengte van de meetwaarden van de ribosomale kaart kort is, kan een meetwaarde vergeleken worden met de verkeerde positie. Daarom is het niet overtuigend om de regio die onder de Ribo-seq valt, simpelweg te beschouwen als de vertaalde regio. Hiertoe, wordt de machine het leren methode gebruikt om de codagecapaciteit van RMRJ te identificeren. Ten eerste haalt CircCode het coderen van RNAs (positieve gegevens) en het noncoderen van RNAs (negatieve gegevens) van een soort van belang uit en gebruikt ze voor modelopleiding door middel van het verschil in kenmerkvectoren tussen het coderen en het noncoderen van RNAs. CircCode gebruikt vervolgens het getrainde model om de Rmrj ‘ s te classificeren die in de vorige stap zijn verkregen door BASiNET. Als RMRJ van circRNA als codage-RNA wordt erkend, dan kan dit circRNA als vertaalde circRNA worden geà dentificeerd.
voorspelling van vertaalde peptiden door Rmrj ‘ s
aangezien de expressie van circRNAs in organismen laag is, tonen de Ribo-seq-gegevens niet duidelijk de exacte 3-nt-periodiciteit in het geval van minder RPFs. Daarom is het moeilijk om de exacte vertaalbeginplaats van een vertaalde circRNA te bepalen. Vanwege de aanwezigheid van een stopcodon in sommige Rmrj ‘ s en omdat het startcodon moeilijk te bepalen is, is de methode om een ORF te vinden op basis van een startcodon en een stopcodon niet haalbaar.
om de ware vertaalgebieden van deze circrna ‘ s te bepalen en het uiteindelijke vertaalproduct te genereren, FragGeneScan (Rho et al., 2010), die eiwit-codeert gebieden in gefragmenteerde genen en genen met frameshifts kan voorspellen, wordt gebruikt om vertaalde peptides te bepalen die door circRNAs worden geproduceerd.
om het omslachtige lopende proces te vermijden, kunnen alle modellen worden aangeroepen door een shell script; de gebruiker kan eenvoudig het gegeven configuratiebestand invullen en het in het script invoeren, en het gehele proces voor het voorspellen van de vertaalde circRNAs zal dan worden uitgevoerd. Bovendien kan CircCode stap voor stap afzonderlijk worden uitgevoerd, zodat de gebruiker de parameters in het midden van de procedure kan aanpassen en de resultaten van elke stap naar wens kan bekijken.
resultaten en discussie
na het testen op meerdere computers bleek CircCode succesvol te draaien met de vereiste afhankelijkheden geïnstalleerd. Om de prestaties van CircCode te testen, gebruikten we gegevens voor mensen en A. thaliana om circRNAs met vertaalpotentieel te voorspellen. De resultaten werden vergeleken met circRNAs die experimenteel als bevestiging zijn geverifieerd. Daarna hebben we de false discovery rate (FDR) waarde van CircCode verder getest. We gebruikten GenRGenS (Ponty et al., 2006) om een gegevensverzameling voor het testen te produceren die op bekende vertaalde circRNAs wordt gebaseerd en bevestigde dat de FDR-waarde binnen een aanvaardbaar bereik en op een laag niveau was. Tot slot evalueerden we het effect van verschillende sequencing diepten van Ribo-seq gegevens op CircCode voorspellingen en vergeleken CircCode met andere software.
vertaalde circRNAs bij mensen en A. thaliana
om de CircCode tool toe te passen op echte data, hebben we eerst de bestanden gedownload, waaronder het menselijk referentie genoom GRCh38, genoomannotatie en menselijk rRNA, van Ensembl. Voor A. thaliana werden de referentie genomen (TAIR10), genoom annotatiebestanden en bijbehorende rRNA-sequenties allemaal gedownload van Ensembl-planten. De Ribo-seq gegevens voor mensen en A. thaliana werden gedownload van RPFdb (toetredingsnummers: GSE96643, GSE81295, GSE88794) (Hsu et al., 2016; Willems et al., 2017), en alle kandidaat circRNAs van mens en A. thaliana werden gedownload van CIRCPedia v2 (Dong et al., 2018) en Plantcirbbase, respectievelijk (Chu et al., 2017). Uiteindelijk identificeerden we 3.610 vertaalde circRNAs van de mens en 1.569 vertaalde circRNAs van A. thaliana met behulp van CircCode (aanvullende gegevens 1).
functionele verrijking van mens en A. thaliana circRNAs met Coderingspotentieel
gebruikmakend van de Circcoderesultaten voor mens en A. thaliana, de online tool KOBAS 3.0 (Wu et al., 2006) werd aangewend om deze vertaalde circRNAs te annoteren op basis van hun oudergenen. Verder voerden we GO (Gene ontologie) functionele analyse en Kegg (Kyoto Encyclopedia of Genes and Genomes) verrijking analyse voor deze vertaalde circRNAs met behulp van de R pakket clusterProfiler (Yu et al., 2012).
de KEGG-resultaten toonden aan dat de menselijke circRNAs verrijkt waren door eiwitverwerking in de endoplasmatische reticulumroute, de koolstofmetabolismeroute en de RNA-transportroute. GO-analyse wees op de deelname van menselijke vertaalde circRNAs in de verordening van molecuulband, ATPase-activiteit, en andere RNA-verbindende biologische processen. Bovendien zijn de vertaalde circRNAs van A. thaliana verrijkt in wegen die verband houden met spanningsweerstand, die suggereren dat zij essentiële rollen in dit proces spelen (aanvullende gegevens 2).
nauwkeurigheidstest voor CircCode
om de nauwkeurigheid van CircCode te onderzoeken, werden testsequenties gegenereerd door GenRGenS, die gebruik maken van het verborgen Markov-model om sequenties te produceren die dezelfde sequentiekenmerken hebben (zoals de frequenties van verschillende nucleotiden, verschillende codons en verschillende nucleotiden aan het begin van de sequentie).
voor deze studie gebruikten we eerder gepubliceerde human translated circRNAs (Yang et al., 2017) als input voor GenRGenS en genereerde 10.000 sequenties om CircCode te testen. We herhaalden de test 10 keer, en gemiddeld werden 27 vertaalde circRNAs elke keer voorspeld. De FDR-waarde werd berekend op 0,0027, wat veel minder is dan 0,05, wat aangeeft dat de voorspelde resultaten geloofwaardig zijn.
bovendien vergeleken we de vertaalde circRNAs van mensen zoals geïdentificeerd door CircCode met geverifieerde polysoom-geassocieerde circRNA-gegevens (Yang et al., 2017). Onder hen werd 60% van de circrna ‘ s geïdentificeerd door CircCode (aanvullende gegevens 3).
invloed van de Ribo-seq Data Sequencing Depth
om de impact van de sequencing depth van Ribo-seq data op de CircCode identificatie resultaten te onderzoeken, hebben we eerst het effect van sequencing depth getest op het aantal vertaalde circrna ‘ s (Figuur 2A). Toen de het rangschikken diepte laag was, was het voorspelde aantal vertaalde circRNAs laag, en het aantal vertaalde circRNAs steeg met stijgende het rangschikken diepte. Het aantal vertaalde circRNAs werd stabiel toen de het rangschikken diepte niet minder dan 10× lineaire transcript dekking bereikte.

Figuur 2 (A) Effect van Ribo-seq gegevens die diepte rangschikken op het voorspelde aantal vertaalde circRNAs. (B) het effect van junction read number (JRN) op Circcodegevoeligheid bij verschillende sequentiedieptes.
ten tweede werd ook de invloed van NMJ op de gevoeligheid bij verschillende sequentiedieptes beoordeeld (figuur 2B). De resultaten toonden aan dat NMJ minder invloed op gevoeligheid had aangezien de het rangschikken diepte toenam. CircCode had ook hogere gevoeligheid wanneer het gebruiken van Ribo-seq gegevens met het hogere rangschikken van diepte.
vergelijking van CircCode met andere Tools
om CircCode te vergelijken met andere tools, zoals CircPro, werd dezelfde set Ribo-seq data (SRR3495999) van A. thaliana gebruikt om vertaalde circna ‘ s te identificeren met behulp van zes processoren, met 16 gigabyte RAM. CircPro identificeerde 44 vertaalde circRNAs in 13 min, terwijl CircCode 76 vertaalde circRNAs in 20 min identificeerde. CircCode is dus gevoeliger dan CircPro op hetzelfde computerhardwareniveau, maar het kost meer tijd. CircPro is beknopt en minder tijdrovend dan CircCode, maar CircCode kan meer circRNAs met codeercapaciteit dan CircPro identificeren.
conclusies
Circrna ’s spelen een belangrijke rol in de biologie en het is van cruciaal belang om circrna’ s met codeervermogen voor volgend onderzoek nauwkeurig te identificeren. Gebaseerd op Python 3, ontwikkelden wij CircCode, een gemakkelijk te gebruiken opdrachtregelhulpmiddel dat hoge gevoeligheid voor het identificeren van vertaalde circRNAs van Ribo-Seq leest met hoge nauwkeurigheid heeft. CircCode vertoont goede prestaties bij zowel planten als dieren. Toekomstige werkzaamheden zullen de stroomafwaartse karakteranalyse aan CircCode toevoegen door elke stap in het proces te visualiseren en de nauwkeurigheid van de voorspelling te optimaliseren.
beschikbaarheid en vereisten
CircCode is beschikbaar op https://github.com/PSSUN/CircCode; besturingssysteem(en): Linux, programmeertalen: Python 3 en R; andere vereisten: bedtools (versie 2.20.0 of hoger), bowtie, STAR, Python 3 pakketten (Biopython, Panda ‘ s, rpy2), R-pakketten (BASiNET, Biostrings). De installatiepakketten voor alle benodigde software zijn beschikbaar op de CircCode homepage. Gebruikers hoeven ze niet individueel te downloaden. De CircCode home page biedt ook gedetailleerde gebruikershandleidingen ter referentie. De tool is vrij beschikbaar. Er zijn geen beperkingen op het gebruik door nonacademics.
Gegevensbeschikbaarheidsverklaring
alle relevante gegevens bevinden zich in het manuscript en de bijbehorende informatiebestanden.
Auteursbijdragen
conceptualisatie: PS, GL. Datacuratie: PS, GL. Formele analyse: PS, GL. Schrijven-originele ontwerp: PS, GL. Schrijven-beoordelen en bewerken: PS, GL.
financiering
dit werk werd ondersteund door subsidies van de National Natural Science Foundation of China (subsidies nrs. 31770333, 31370329 en 11631012), het programma voor excellente talenten in de nieuwe eeuw in de Universiteit (NCET-12-0896), en de fondsen voor fundamenteel onderzoek voor de centrale universiteiten (nr. GK201403004). De financieringsinstanties hadden geen rol in de studie, de opzet ervan, de gegevensverzameling en-analyse, het besluit tot publicatie of de voorbereiding van het manuscript. De financiers hadden geen rol in het ontwerp van de studie, het verzamelen en analyseren van gegevens, het besluit tot publicatie of de voorbereiding van het manuscript.
belangenconflict
de auteurs verklaren dat het onderzoek werd uitgevoerd zonder enige commerciële of financiële relatie die als een potentieel belangenconflict kon worden opgevat.
aanvullend materiaal
het aanvullende materiaal voor dit artikel is online te vinden op: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
aanvullende gegevens 1 / de volgorde van de voorspelde vertaalde circRNA en korte peptide.
aanvullende gegevens 2 / GO verrijking en Kegg verrijking resultaten voor mensen en Arabidopsis thaliana.
aanvullende gegevens 3 / vergelijking van voorspelde vertaalde circRNAs met gevalideerde vertaalde circRNAs.
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmatic: een flexibele trimmer voor illumina sequentiegegevens. Bioinformatics 30, 2114-2120. doi: 10.1093 / bioinformatics / btu170
PubMed Abstract / CrossRef Full Text / Google Scholar
Brar, G. A., Weissman, J. S. (2015). Het ribosoomprofileren onthult wat, wanneer, waar en hoe van eiwitsynthese. Nat. Rev. Mol. Cel Biol. 16, 651–664. doi: 10.1038 / nrm4069
PubMed Abstract / CrossRef Full Text / Google Scholar
Chen, L.-L., Yang, L. (2015). Regulatie van circRNA biogenese. RNA Biol. 12, 381–388. doi: 10.1080/15476286.2015.1020271
PubMed Abstract / CrossRef Full Text / Google Scholar
Chu, Q., Zhang, X., Zhu, X., Liu, C., Mao, L., Ye, C., et al. (2017). PlantcircBase: een database voor plant circulaire RNAs. Mol. Plant 10, 1126-1128. doi: 10.1016 / j.molp.2017.03.003
PubMed Abstract / CrossRef Full Text / Google Scholar
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). Ster: ultrasnelle universele RNA-seq aligner. Bioinformatica 29, 15-21. doi: 10.1093 / bioinformatics / bts635
PubMed Abstract / CrossRef Full Text / Google Scholar
Dong, R., Ma, X.-K., Chen, L.-L., Yang, L. (2019). “Genome-wide annotation of circRNAs and their alternative back-splicing / splicing with CIRCexplorer Pipeline,” in Epitranscriptomics. EDS. Wajapeyee, N., Gupta, R. (New York, NY: Springer New York), 137-149. doi: 10.1007/978-1-4939-8808-2_10
CrossRef Full Text / Google Scholar
Dong, R., Ma, X.-K., Li, G.-W., Yang, L. (2018). CIRCpedia v2: een bijgewerkte database voor uitgebreide circulaire RNA annotatie en expressie vergelijking. Genomics Proteomics Bioinf. 16, 226–233. doi: 10.1016 / j.gpb.2018.08.001
CrossRef Full Text / Google Scholar
Gao, Y., Wang, J., Zhao, F. (2015). CIRI: een efficiënt en onbevooroordeeld algoritme voor de novo circulaire RNA-identificatie. Genome Biol. 16, 4. doi: 10.1186 / s13059-014-0571-3
PubMed Abstract / CrossRef Full Text / Google Scholar
Guttman, M., Russell, P., Ingolia, N. T., Weissman, J. S., Lander, E. S. (2013). Ribosoom het profileren verstrekt bewijsmateriaal dat de grote noncoding RNAs geen proteã nen coderen. Cel 154, 240-251. doi: 10.1016 / j.cel.2013.06.009
PubMed Abstract / CrossRef Full Text / Google Scholar
Hsu, P. Y., Benfey, P. N. (2018). Klein maar machtig: functionele peptiden gecodeerd door kleine ORFs in installaties. PROTEOMICS 18, 1700038. doi: 10.1002 / pmic.201700038
CrossRef Full Text / Google Scholar
Hsu, P. Y., Calviello, L., Wu, H.-Y. L., Li, F.-W., Rothfels, C. J., Ohler, U., et al. (2016). Super-resolutie ribosoom profilering onthult niet-geannoteerde vertaalgebeurtenissen in Arabidopsis. Proc. Natl. Acad. Sci. 113, E7126-E7135. doi: 10.1073 / pnas.1614788113
CrossRef Full Text / Google Scholar
Ingolia, N. T., Lareau, L. F., Weissman, J. S. (2011). Ribosoom profilering van muis embryonale stamcellen onthult de complexiteit en dynamiek van zoogdierproteomes. Cel 147, 789-802. doi: 10.1016 / j.cel.2011.10.002
PubMed Abstract / CrossRef Full Text / Google Scholar
Ito, E. A., Katahira, I., Vicente, F. F., da, R., Pereira, L. F. P., Lopes, F. M. (2018). BASiNET-BiologicAl Sequences NETwork: een gevalstudie over het coderen en niet-coderen RNAs identificatie. Nucleïnezuren res.46, e96–e96. doi: 10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093 / bioinformatics / bty948
CrossRef Full Text / Google Scholar
Kearse, M. G., Wilusz, J. E. (2017). Non-AUG vertaling: een nieuwe start voor eiwitsynthese in eukaryoten. Genes Dev. 31, 1717–1731. doi: 10.1101/gad.305250.117
PubMed Abstract / CrossRef Full Text / Google Scholar
Kong, L., Zhang, Y., Ye, Z.-Q., Liu, X.-Q., Zhao, S.-Q., Wei, L., et al. (2007). CPC: beoordeel het eiwit-codeert potentieel van afschriften gebruikend opeenvolgingseigenschappen en steun vectormachine. Nucleïnezuren Res. 35, W345-W349. doi: 10.1093/nar / gkm391
PubMed Abstract / CrossRef Full Text / Google Scholar
Langmead, B., Salzberg, S. L. (2012). Snelle gapped-lezen uitlijning met vlinderdas 2. Nat. Methods 9, 357-359. doi: 10.1038 / nmeth.1923
PubMed Abstract / CrossRef Full Text / Google Scholar
Meng, X., Chen, Q., Zhang, P., Chen, M. (2017). CircPro: een geïntegreerd hulpmiddel voor de identificatie van circRNAs met eiwit-codeert potentieel. Bioinformatics 33, 3314-3316. doi: 10.1093 / bioinformatics / btx446
PubMed Abstract / CrossRef Full Text / Google Scholar
Michel, A. M., Baranov, P. V. (2013). Ribosoom profilering: een Hi-Def monitor voor eiwitsynthese op de genoombrede schaal: ribosoom profilering. Wiley Interdiscip. Rev. RNA 4, 473-490. doi: 10.1002 / wrna.1172
PubMed Abstract / CrossRef Full Text / Google Scholar
Pamudurti, N. R., Bartok, O., Jens, M., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., et al. (2017). Vertaling van CircRNAs. Mol. Cel 66, 9-21.e7. doi: 10.1016 / j.molcel.2017.02.021
PubMed Abstract / CrossRef Full Text / Google Scholar
Ponty, Y., Termier, M., Denise, A. (2006). GenRGenS: software voor het genereren van willekeurige genomische sequenties en structuren. Bioinformatica 22, 1534-1535. doi: 10.1093 / bioinformatics / btl113
PubMed Abstract / CrossRef Full Text / Google Scholar
Rho, M., Tang, H., Ye, Y. (2010). FragGeneScan: het voorspellen van genen in korte en foutgevoelige leest. Nucleic Acids res. 38, e191-e191. doi: 10.1093/nar / gkq747
PubMed Abstract / CrossRef Full Text / Google Scholar
Slavoff, S. A., Mitchell, A. J., Schwaid, A. G., Cabili, M. N., Ma, J., Levin, J. Z., et al. (2013). Peptidomic ontdekking van korte open lezingskader-gecodeerde peptides in menselijke cellen. Nat. Scheikunde. Biol. 9, 59–64. doi: 10.1038 / nchembio.1120
PubMed Abstract / CrossRef Full Text / Google Scholar
Spealman, P., Naik, A. W., May, G. E., Kuersten, S., Freeberg, L., Murphy, R. F., et al. (2018). Behouden niet-AUG uORFs onthuld door een nieuwe regressieanalyse van ribosoom profilering gegevens. Genome Res. 28, 214-222. doi: 10.1101 / gr.221507.117
PubMed Abstract / CrossRef Full Text / Google Scholar
Vicens, Q., Westhof, E. (2014). Biogenese van circulaire RNAs. Cel 159, 13-14. doi: 10.1016 / j.cel.2014.09.005
PubMed Abstract / CrossRef Full Text / Google Scholar
Willems, P., Ndah, E., Jonckheere, V., Stael, S., Sticker, A., Martens, L., et al. (2017). N-terminal proteomics assisteerde de profilering van het onontgonnen vertaalinitiatielandschap in Arabidopsis thaliana. Mol. Cel. Proteomics 16, 1064-1080. doi: 10.1074 / mcp.M116. 066662
PubMed Abstract / CrossRef Full Text / Google Scholar
Wu, J., Mao, X., Cai, T., Luo, J., Wei, L. (2006). KOBAS server: een web-based platform voor geautomatiseerde annotatie en pathway identificatie. Nucleïnezuren Res.34, W720–W724. doi: 10.1093/nar / gkl167
PubMed Abstract / CrossRef Full Text / Google Scholar
Yang, L., Fu, J., Zhou, Y. (2018). Circulaire RNAs en hun opkomende rollen in immuunregulatie. Voorkant. Immunol. 9, 2977. doi: 10.3389 / fimmu.2018.02977
PubMed Abstract / CrossRef Full Text / Google Scholar
Yang, Y., Fan, X., Mao, M., Song, X., Wu, P., Zhang, Y., et al. (2017). Uitgebreide vertaling van circulaire RNAs gedreven door N6-methyladenosine. Cel Res. 27, 626-641. doi: 10.1038 / cr.2017.31
PubMed Abstract / CrossRef Full Text / Google Scholar
Yu, G., Wang, L.-G., Han, Y., He, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar