Con la lesión de tejido sano, se desarrolla una progresión predecible de eventos fisiológicos. Esta progresión se puede dividir en las fases de inflamación, proliferación y maduración. Cada fase se caracteriza por la elaboración secuencial de citocinas distintivas por células específicas. Vea las imágenes a continuación.
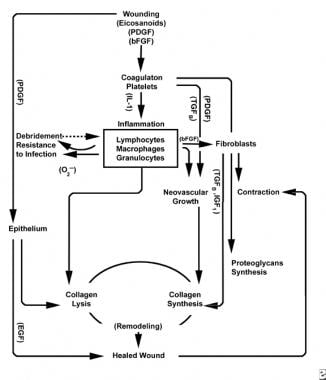 Esquemas del proceso de cicatrización de heridas.
Esquemas del proceso de cicatrización de heridas. 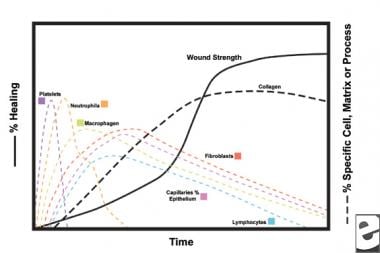 las características Celulares del proceso de cicatrización de heridas.
las características Celulares del proceso de cicatrización de heridas. La fase inflamatoria
La fase inflamatoria lanza simultáneamente mecanismos y vías hemostáticas que crean los signos cardinales clínicamente reconocibles de inflamación: rubor (enrojecimiento), calor (calor), tumor (hinchazón), dolor (dolor) y functio laesa (pérdida de función).
La lesión en el tejido vascular inicia la cascada de coagulación extrínseca al liberar calcio intracelular y factor tisular que activan el factor VII. El tapón de fibrina resultante logra hemostasia con la ayuda de una vasoconstricción refleja. Este tapón actúa como una red para la agregación de plaquetas, el tipo celular más común y «característico» de la fase inflamatoria temprana.
Las plaquetas elaboran una serie de sustancias proinflamatorias, como el difosfato de adenosina, el factor de crecimiento tisular beta (TGF-ß) y los factores de crecimiento derivados de plaquetas (PDGF). Estos factores de crecimiento actúan sobre las células circundantes y estimulan la quimiotaxis de neutrófilos, monocitos y fibroblastos en el área lesionada.
Los tejidos lesionados, a través de la fosfolipasa A activada, catalizan simultáneamente los ácidos araquidónicos para producir prostaglandinas vasoactivas y tromboxano, conocidos colectivamente como eicosanoides. Los eicosanoides median en la actividad que influye en la formación del tapón plaquetario, la permeabilidad vascular y la quimiotaxis celular para influir en la cicatrización de heridas. Por ejemplo, el tromboxano A2 media la vasoconstricción y la agregación plaquetaria.
Después de la vasoconstricción inicial, los signos clásicos de inflamación se manifiestan por el aumento de la permeabilidad vascular. Rubor es el resultado de la vasodilatación, mediada por prostaciclina (PGI2), prostaglandina A (PGA), prostaglandina D (PGD) y prostaglandina E (PGE). El tumor y el calor se desarrollan a medida que las brechas endoteliales vasculares se agrandan, lo que permite la salida de proteína plasmática y líquido al espacio intersticial. Estos cambios son potenciados por la PGE2 y la prostaglandina F2a (PGF2a) y permiten la entrada de células inflamatorias en el área de la lesión, incluidas las células que se elaboran. El dolor se detecta cuando PGI2, PGE y PGE2 actúan sobre los nociceptores periféricos.
En la segunda etapa de la fase inflamatoria, los leucocitos suplantan a las plaquetas como el tipo de célula dominante, atraídos por la quimiotaxis. Los glóbulos blancos (glóbulos blancos) son las células predominantes durante los primeros 3 días después de la lesión; su número alcanza su punto máximo a las 48 horas aproximadamente. Los polimorfonucleocitos (PMNs) son los primeros en iniciar actividades bactericidas utilizando mediadores inflamatorios y metabolitos de radicales libres de oxígeno. Sin embargo, la cicatrización normal de heridas puede ocurrir sin PMNs. Otro leucocito, el linfocito T auxiliar, elabora la interleucina – 2 (IL–2). La IL-2 promueve una mayor proliferación de células T para aumentar la respuesta inmunogénica a la lesión.
A medida que los leucocitos PMN comienzan a disminuir después de 24-36 horas, los monocitos circulantes ingresan a la herida y maduran en macrófagos de tejido. Estas células desbridan la herida a nivel microscópico y producen una amplia variedad de sustancias importantes, como la IL-1 y el factor de crecimiento básico de fibroblastos (bFGF). La IL-1 estimula la proliferación de células inflamatorias y promueve la angiogénesis a través de la replicación de células endoteliales. El bFGF es un factor quimiotáctico y mitogénico para los fibroblastos y las células endoteliales. A diferencia del PMNs, la depleción de macrófagos afecta gravemente la cicatrización de heridas, ya que el desbridamiento, la proliferación de fibroblastos y la angiogénesis disminuyen.
Hacia el final del ciclo inflamatorio, el entorno evolutivo de los eicosanoides en la herida interactúa con los tipos de células presentes, lo que resulta en la síntesis de fibroblastos de colágeno y sustancia molida (a partir de una mayor proporción de PGF2a a PGE2). Además, los factores de crecimiento derivados de los macrófagos se encuentran ahora en niveles óptimos, lo que influye fuertemente en la entrada de fibroblastos y luego de queratinocitos y células endoteliales en la herida. A medida que las células mononucleares continúan reemplazando los glóbulos blancos y los macrófagos, comienza la fase proliferativa.
La fase proliferativa
Dos o tres días después de la herida, los fibroblastos migran hacia adentro desde los márgenes de la herida sobre la matriz fibrinosa establecida durante la fase inflamatoria. Durante la primera semana, los fibroblastos comienzan a producir glicosaminoglicanos y proteoglicanos, la sustancia fundamental para el tejido de granulación, así como colágeno, en respuesta al bFGF y TGF-ß sintetizados en macrófagos, así como al PDGF.
Los fibroblastos pronto se convierten en el tipo de célula dominante, con un pico de 1 a 2 semanas. Generan no solo moléculas de colágeno, sino también citocinas como PDGF , TGF-ß, bFGF, factor de crecimiento de queratinocitos y factor de crecimiento insulínico-1. Los fibroblastos también ensamblan moléculas de colágeno en fibras, que están reticuladas y organizadas en haces. El colágeno es el componente principal del tejido conectivo agudo de la herida, y la producción neta continúa durante las próximas 6 semanas. El aumento del contenido de colágeno de la herida se correlaciona con el aumento de la resistencia a la tracción.
Los queratinocitos y las células endoteliales también proliferan durante este tiempo, produciendo eventualmente factores de crecimiento autocrinos que mantienen su crecimiento. La expansión endotelial contribuye a la angiogénesis, ya que los vasos intactos generan brotes en el tejido de granulación. La neovascularización facilita el crecimiento de la línea de avance de fibroblastos en la herida, proporcionándoles los nutrientes y citoquinas necesarios.
La degradación del coágulo de fibrina y la matriz provisional se acompaña de la deposición de tejido de granulación (sustancia molida, colágeno, capilares), que continúa hasta que se cubre la herida. La disminución de los niveles de ácido hialurónico (en sustancia molida) y el aumento de los niveles de sulfato de condroitina ralentizan la migración y proliferación de fibroblastos, al tiempo que inducen la diferenciación de fibroblastos, pasando a la fase de maduración de la cicatrización de heridas.
La fase de maduración
Durante las primeras 6 semanas, la producción de colágeno nuevo domina el proceso de cicatrización de heridas, depositado aleatoriamente en el tejido de granulación de heridas agudas. A medida que la herida madura, el colágeno se remodela en una estructura más organizada con mayor resistencia a la tracción. Gradualmente, el colágeno tipo I reemplaza al tipo III hasta que se alcanza la proporción normal de piel de 4:1. A medida que continúa la remodelación, la colagenólisis de metaloproteinasa de matriz logra un estado estacionario con la síntesis de colágeno. Mesetas de resistencia a la tracción al 80% de la resistencia original aproximadamente 1 año después del juramento.
Superficiales a esta actividad, las células epiteliales continúan migrando hacia adentro desde el borde de la herida hasta que el defecto está cubierto. En este punto, la inhibición por contacto induce la transformación de fibroblastos en miofibroblastos, que contienen fibras contráctiles de actina. La contracción de la herida sigue, reemplazando el volumen de tejido lesionado con tejido nuevo, aunque el papel exacto del miofibroblasto no se ha dilucidado completamente.
Los elementos disuasorios para la cicatrización de heridas
Las heridas agudas generalmente se realizan a través de un proceso reparador ordenado y oportuno que resulta en una restauración duradera de la integridad anatómica y funcional. Sin embargo, varios factores fisiológicos y mecánicos pueden afectar la respuesta de curación, lo que resulta en una herida crónica que no avanza a través de la progresión gradual habitual. La infección local, la hipoxia, el trauma, los cuerpos extraños o los problemas sistémicos como la diabetes mellitus, la desnutrición, la inmunodeficiencia o los medicamentos son los responsables más frecuentes.
Todas las heridas están contaminadas, pero con mayor éxito resisten la infección invasiva. Cuando la concentración supera los 100.000 (105) organismos por gramo de tejido o el sistema inmunitario se ve comprometido, con frecuencia se produce una infección. La celulitis prolonga la fase inflamatoria al mantener altos niveles de citocinas proinflamatorias y proteasas tisulares, que degradan el tejido de granulación y los factores de crecimiento tisular, y al retrasar la deposición de colágeno.
El desbridamiento (quirúrgico, enzimático y / o mediante cambios de vendaje) y los antibióticos son los pilares del tratamiento con antibióticos. El desbridamiento elimina el tejido desvitalizado, que puede ser una fuente de endotoxinas que inhiben la migración de fibroblastos y queratinocitos a la herida. Los cuerpos extraños también pueden requerir la eliminación, ya que la presencia de una sutura de seda reduce el número de bacterias necesarias para incitar a la infección 10,000 veces. (Para obtener una descripción detallada de la técnica, consulte el artículo de Referencia de Medscape Extracción de cuerpos extraños en heridas.)
La hipoxia celular retrasa la cicatrización de heridas por diversos medios. La reticulación de las fibras de colágeno requiere oxígeno para hidroxilar prolina y lisina y falla cuando la presión tisular está por debajo de 40 mm Hg. La potencia bactericida de la fosforilación oxidativa de leucocitos también sufre en un entorno hipóxico, reduciendo el umbral de infección. Las medidas para mejorar el suministro de oxígeno dependen de la etiología. El consumo de tabaco, que causa vasoconstricción y aumenta la adherencia a las plaquetas, debe interrumpirse. La angioplastia o el injerto de bypass arterial pueden ser necesarios para la enfermedad vascular periférica. Se pueden indicar medidas complementarias para mejorar la perfusión sistémica en casos de insuficiencia cardíaca. Se debe tratar un valor de hematocrito inferior al 15% y restaurar la euvolemia, según sea necesario. La estasis venosa o la insuficiencia linfática pueden mejorarse con prendas compresivas.
La enfermedad sistémica puede prolongar o interrumpir drásticamente la cicatrización de heridas. La glicosilación en la diabetes mellitus perjudica la fagocitosis bacteriana de neutrófilos y macrófagos, prolongando la fase inflamatoria. La fase proliferativa también se prolonga en la misma enfermedad a medida que los eritrocitos se vuelven menos flexibles y menos capaces de suministrar oxígeno a la herida para el metabolismo de los tejidos y la síntesis de colágeno.
La desnutrición resulta en una disminución de la proliferación de fibroblastos, un deterioro de la neovascularización y una disminución de la inmunidad celular y humoral. Las heridas ejercen mayores demandas metabólicas, particularmente dentro del tejido de granulación. Los aminoácidos, como la metionina, la prolina, la glicina y la lisina, son esenciales para el funcionamiento normal de las células y la reparación de heridas cutáneas. Los ácidos grasos son constituyentes críticos de las membranas celulares y son el sustrato de los eicosanoides que median el proceso inflamatorio. Los ácidos grasos esenciales linolénico y ácido linoleico deben suministrarse en la dieta, ya que el cuerpo humano es incapaz de la síntesis de novo de estas moléculas.
Deben estar disponibles vitaminas y minerales adecuados para el metabolismo celular, que actúan como señales celulares y cofactores. La vitamina C (ácido ascórbico) y el hierro son necesarios para la hidroxilación de la lisina y la prolina, que se entrecruzan y estabilizan la estructura de triple hélice del colágeno; el cobre también desempeña un papel en la estabilización del colágeno. La vitamina A (ácido retinoico) desempeña un papel importante en la modulación de la producción y degradación del colágeno y es particularmente importante en la epitelización. Un potente antioxidante, la vitamina E (alfa tocoferol) parece acelerar la cicatrización dérmica y ósea en los animales, y la suplementación puede tener un papel en los seres humanos. La deficiencia de trazas de metal, en particular de zinc, también está asociada con una mala cicatrización de heridas; esto debe reponerse, según corresponda.
Ovidio supuestamente escribió: «los medicamentos a veces sanan, a veces matan.»Esto es ciertamente cierto con respecto a la curación de heridas. Los corticosteroides embotan los procesos de toda la fase inflamatoria. La vitamina A (por vía tópica o 25.000 UI/d por vía oral) mitiga los efectos curativos perjudiciales de los corticosteroides, pero la hepatotoxicidad puede resultar del uso prolongado (es decir, >1 mes). Los medicamentos antiinflamatorios no esteroideos (AINE) también interfieren con el metabolismo del ácido araquidónico y, por lo tanto, con la cicatrización de heridas. Además, los AINE inhiben la función plaquetaria, uno de los primeros procesos en la fase inflamatoria.
Un estudio de Sutcliffe et al sugirió que la regulación ascendente de la conexina de la proteína de unión de brecha es común en heridas crónicas. Al examinar connexin en tres tipos de heridas (pierna venosa, pie diabético y úlceras por presión), los investigadores encontraron que cada tipo de herida mostraba una regulación ascendente de la conexina epidérmica 43, la conexina 26 y la conexina 30, así como de la conexina dérmica 43.