- Introducción
- Métodos
- Filtrado de Datos de perfiles Ribosómicos
- Ensamblar genomas virtuales
- Determinación de la Región Mapeada de Lectura de Ribo-seq en una Unión (RMRJ) de circRNAs
- Entrenamiento del Modelo y Clasificación de RMRJs
- La predicción de péptidos Traducidos por RMRJs
- Resultados y discusión
- circRNAs traducidos en Humanos y A. thaliana
- Enriquecimiento funcional de los circRNAs Humanos y A. thaliana Con Potencial de codificación
- Prueba de precisión para CircCode
- Influencia de la Profundidad de Secuenciación de Datos Ribo-seq
- Comparación de CircCode Con Otras Herramientas
- Conclusiones
- Disponibilidad y requisitos
- Declaración de disponibilidad de datos
- Contribuciones de autor
- Financiación
- Conflicto de intereses
- Material suplementario
Introducción
Los ARN circulares (ARN) son un tipo especial de molécula de ARN no codificante que se ha convertido en un tema de investigación candente en el campo del ARN y está recibiendo mucha atención (Chen y Yang, 2015). En comparación con los ARN lineales tradicionales (que contienen extremos de 5′ y 3′), las moléculas de ARN circular generalmente tienen una estructura circular cerrada, lo que las hace más estables y menos propensas a la degradación (Vicens y Westhof, 2014). Aunque la existencia de circRNAs se conoce desde hace algún tiempo, estas moléculas se consideraban un subproducto del empalme de ARN. Sin embargo, con el desarrollo de tecnologías bioinformáticas y de secuenciación de alto rendimiento, los circRNAs se han vuelto ampliamente reconocidos en animales y plantas (Chen y Yang, 2015). Estudios recientes también han demostrado que un gran número de circRNAs se pueden traducir en pequeños péptidos en las células (Pamudurti et al., 2017) y tienen roles clave a pesar de su nivel de expresión a veces bajo (Hsu y Benfey, 2018; Yang et al., 2018). Aunque se está identificando un número cada vez mayor de circRNAs, sus funciones en plantas y animales generalmente quedan por estudiar. Además de sus funciones como señuelos de miRNA, los circRNAs tienen un importante potencial de traducción, pero no hay herramientas disponibles para predecir específicamente las capacidades de traducción de estas moléculas (Jakobi y Dieterich, 2019).
Existen varias herramientas para la predicción e identificación de circRNAs, como CIRI (Gao et al., 2015), CIRCexplorer (Dong et al., 2019), CircPro (Meng et al., 2017), y circtools (Jakobi et al., 2018). Entre ellos, CircPro puede revelar circRNAs traducidos calculando una puntuación potencial de traducción para circRNAs basada en la CCP (Kong et al., 2007), que es una herramienta para identificar el marco de lectura abierto (ORF) en una secuencia dada. Sin embargo, debido a que algunos circRNAs no utilizan el codón de inicio durante la traducción (Ingolia et al., 2011; Slavoff et al., 2013; Kearse y Wilusz, 2017; Spealman et al., 2018), el empleo de CPC puede filtrar algunos circRNAs verdaderamente traducidos. En este estudio, utilizamos BASiNET (BAS et al., 2018), que es un clasificador de ARN basado en los métodos de aprendizaje automático (modelo random forest y J48). Inicialmente transforma los ARN de codificación dados (datos positivos) y los ARN no codificantes (datos negativos) y los representa como redes complejas; luego extrae las medidas topológicas de estas redes y construye un vector de características para entrenar el modelo que se utiliza para clasificar la capacidad de codificación de los ARN circulares. Con este método, se evita el filtrado erróneo de circRNAs traducidos que no son iniciados por AUG. Además, la tecnología Ribo-seq, que se basa en la secuenciación de alto rendimiento para monitorear RPF (fragmentos protegidos ribosómicos) de transcripciones (Guttman et al., 2013; Brar y Weissman, 2015), se puede utilizar para determinar la ubicación de los circRNAs que se están traduciendo (Michel y Baranov, 2013). Para identificar la capacidad de codificación de los circRNAs, desarrollamos la herramienta CircCode, que involucra un marco basado en Python 3, y aplicamos CircCode para investigar el potencial de traducción de los circRNAs de humanos y Arabidopsis thaliana. Nuestro trabajo proporciona un rico recurso para estudiar más a fondo las funciones de los circRNAs con capacidad de codificación.
Métodos
El código fue escrito en el lenguaje de programación Python 3; utiliza Trimmomatic (Bolger et al., 2014), pajarita (Langmead y Salzberg, 2012) y ESTRELLA (Dobin et al., 2013) para filtrar las lecturas de Ribo-seq en bruto y mapear estas lecturas filtradas al genoma. CircCode luego identifica regiones de lectura mapeadas Ribo-seq en círculos que contienen uniones. Después de eso, las secuencias mapeadas candidatas en los circRNAs se ordenan en base a clasificadores (modelo J48) en ARN codificados y ARN no codificados por BASiNET. Finalmente, los péptidos cortos producidos por la traducción se identifican como regiones codificantes potenciales de los circRNAs. Todo el proceso de CircCode consta de cinco pasos (Figura 1).FIGURA
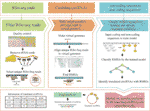
Figura 1 El flujo de trabajo de CircCode. La capa superior representa el archivo de entrada requerido para cada paso del código. La capa intermedia se divide en tres partes, y cada parte representa una etapa diferente de operación. De izquierda a derecha, la primera parte representa el filtrado de los datos Ribo-seq; el control de calidad es ejecutado por Trimmatic, y las lecturas de ARNr son eliminadas por pajarita. La segunda parte representa los pasos utilizados para producir el genoma virtual y alinear las lecturas filtradas con el genoma virtual con STAR. La última parte representa la identificación de los círculos traducidos por aprendizaje automático. La capa inferior representa el último paso utilizado para predecir los péptidos traducidos de los circRNAs y los resultados finales de salida, incluida la información sobre los circRNAs traducidos y sus productos de traducción.
Filtrado de Datos de perfiles Ribosómicos
En primer lugar, Trimmomatic elimina fragmentos y adaptadores de baja calidad en las lecturas Ribo-Seq con los parámetros predeterminados para obtener lecturas Ribo-seq limpias. En segundo lugar, estas lecturas limpias de Ribo-seq se asignan a una biblioteca de ARNr para eliminar las lecturas derivadas de ARNr usando pajarita. Debido a que las longitudes de lectura de Ribo-seq son relativamente cortas (generalmente menos de 50 pb), es posible que una lectura coincida con varias regiones. En este caso, es difícil determinar a qué región corresponde una lectura en particular. Para evitar esto, las lecturas limpias de Ribo-seq se asignan al genoma de una especie de interés, y las lecturas que no están perfectamente alineadas con el genoma se consideran las lecturas finales únicas de Ribo-seq.
Ensamblar genomas virtuales
Los circRNAs generalmente aparecen como moléculas en forma de anillo en eucariotas, y se pueden identificar en función de sus uniones de empalme posterior. Sin embargo, las secuencias de circRNAs en el archivo fasta son a menudo en forma lineal. En teoría, el resultado indica que la unión está entre el nucleótido terminal de 5′ y el nucleótido terminal de 3′, aunque la unión y la secuencia cerca de la unión no se pueden ver directamente, alineando así las lecturas de Ribo-seq con secuencias de ARNc, incluidas las uniones, de una manera directa.
CircCode conecta la secuencia de cada circRNA en tándem de tal manera que la unión para cada uno está en el medio de la secuencia recién construida. También separamos cada unidad de serie por 100 nucleótidos N para evitar confusiones en el paso de alineación de la secuencia (la longitud de cada RPF es inferior a 50 pb). Finalmente, obtuvimos un genoma virtual que consistía solo en circRNAs candidatos en tándem separados por 100 Ns. Debido a que CircCode se centra solo en la alineación entre las lecturas de Ribo-seq y las secuencias de ARNc, podemos investigar el potencial de codificación de los ARNc mapeando las lecturas de Ribo-seq a este genoma virtual, lo que puede ahorrar una gran cantidad de tiempo computacional (el genoma virtual es mucho más pequeño que el genoma completo) y aumentar la precisión (evitando la interferencia entre las comparaciones de secuencias aguas arriba y aguas abajo de los ARNC).
Determinación de la Región Mapeada de Lectura de Ribo-seq en una Unión (RMRJ) de circRNAs
Las lecturas únicas finales de Ribo-seq se asignan a un genoma virtual creado previamente utilizando STAR. Debido a que cada unidad de circRNA en tándem estaba separada por 100 bases N antes de producir el genoma virtual, la longitud de intrón más grande se estableció para no exceder las 10 bases con el parámetro «–alignIntronMax 10.»Este parámetro elimina cualquier interacción entre diferentes circRNAs en la alineación de secuencias. En el segundo paso de la producción del genoma virtual, CircCode almacena información de unión posicional para cada circRNA en el genoma virtual. Si la región mapeada de lectura de Ribo-seq en el genoma virtual incluye la unión del circRNA, y el número de lecturas mapeadas de Ribo-seq en la unión (NMJ) es mayor que 3, la región mapeada de lectura de Ribo-seq en la unión de los circRNAs puede considerarse como un RMRJ, que revela un segmento aproximadamente traducido de circRNAs cerca del sitio de la unión.
Entrenamiento del Modelo y Clasificación de RMRJs
Aunque RMRJs puede constituir una prueba poderosa de traducción, todavía hay algunas deficiencias en este método. Debido a que la longitud de las lecturas del mapa ribosómico es corta, una lectura puede compararse con la posición incorrecta. Por lo tanto, no es convincente considerar simplemente la región cubierta por la lectura Ribo-seq como la región traducida. Con este fin, se utiliza el método de aprendizaje automático para identificar la capacidad de codificación del RMRJ. En primer lugar, CircCode extrae ARN codificantes (datos positivos) y ARN no codificantes (datos negativos) de una especie de interés y los utiliza para el entrenamiento de modelos por medio de la diferencia en vectores de características entre ARN codificantes y no codificantes. CircCode luego utiliza el modelo entrenado para clasificar los RMRJ obtenidos en el paso anterior por BASiNET. Si el RMRJ de un circRNA se reconoce como ARN codificador, entonces este circRNA se puede identificar como un circRNA traducido.
La predicción de péptidos Traducidos por RMRJs
Como la expresión de circRNAs en los organismos es baja, los datos de Ribo-seq no muestran claramente la periodicidad exacta de 3-nt en el caso de menos FPR. Por lo tanto, es difícil determinar el sitio exacto de inicio de traducción de un circRNA traducido. Debido a la presencia de un codón de parada en algunos RMRJ y debido a que el codón de inicio es difícil de determinar, el método de encontrar un ORF basado en un codón de inicio y un codón de parada no es factible.
Para determinar las regiones de traducción verdaderas de estos círculos y generar el producto de traducción final, FragGeneScan(Rho et al., 2010), que puede predecir regiones codificantes de proteínas en genes fragmentados y genes con cambios de marco, se utiliza para determinar los péptidos traducidos producidos por los circRNAs.
Para evitar el engorroso proceso de ejecución, todos los modelos pueden ser llamados por un script de shell; el usuario simplemente puede completar el archivo de configuración dado e ingresarlo en el script, y se ejecutará todo el proceso para predecir los circRNAs traducidos. Además, CircCode se puede ejecutar por separado, paso a paso, de modo que el usuario pueda ajustar los parámetros en medio del procedimiento y ver los resultados de cada paso como desee.
Resultados y discusión
Después de realizar pruebas en varios equipos, se encontró que CircCode se ejecutaba correctamente con las dependencias necesarias instaladas. Para probar el rendimiento de CircCode, utilizamos datos para humanos y A. thaliana para predecir circRNAs con potencial de traducción. Los resultados se compararon con circRNAs que se han verificado experimentalmente como confirmación. A partir de entonces, probamos el valor de tasa de descubrimiento falso (FDR) de CircCode más adelante. Usamos GenRGenS (Ponty et al., 2006) para generar un conjunto de datos para pruebas basado en circRNAs traducidos conocidos y confirmó que el valor FDR estaba dentro de un rango aceptable y a un nivel bajo. Finalmente, evaluamos el efecto de las diferentes profundidades de secuenciación de los datos Ribo-seq en las predicciones de CircCode y comparamos CircCode con otro software.
circRNAs traducidos en Humanos y A. thaliana
Para aplicar la herramienta CircCode a datos reales, primero descargamos los archivos que incluyen el genoma humano de referencia GRCh38, la anotación del genoma y el ARNr humano, de Ensembl. Para A. thaliana, los genomas de referencia (TAIR10), los archivos de anotación del genoma y las secuencias de ARNr correspondientes se descargaron de Plantas Ensembl. Los datos de Ribo-seq para humanos y A. thaliana se descargaron de RPFdb (números de acceso: GSE96643, GSE81295, GSE88794) (Hsu et al., 2016; Willems et al., 2017), y todos los circRNAs candidatos de human y A. thaliana se descargaron de Circipedia v2 (Dong et al., 2018) y Plantcirbase, respectivamente (Chu et al., 2017). Finalmente, identificamos 3.610 circRNAs traducidos de humanos y 1.569 circRNAs traducidos de A. thaliana utilizando el código CircCode (Datos suplementarios 1).
Enriquecimiento funcional de los circRNAs Humanos y A. thaliana Con Potencial de codificación
Utilizando los resultados de CircCode para humanos y A. thaliana, la herramienta en línea KOBAS 3.0 (Wu et al., 2006) se empleó para anotar estos circRNAs traducidos en función de sus genes padres. Además, realizamos análisis funcionales de GO (Gene Ontology) y análisis de enriquecimiento de KEGG (Kyoto Encyclopedia of Genes and Genomes) para estos circRNAs traducidos utilizando el paquete R clusterProfiler (Yu et al., 2012).
Los resultados de KEGG mostraron que los ARN humanos estaban enriquecidos en el procesamiento de proteínas en la vía del retículo endoplásmico, la vía del metabolismo del carbono y la vía de transporte del ARN. El análisis de GO indicó la participación de circRNAs traducidos humanos en la regulación de la unión de moléculas, la actividad de la ATPasa y otros procesos biológicos relacionados con el empalme de ARN. Además, los circRNAs traducidos de A. thaliana se enriquecen en vías relacionadas con la resistencia al estrés, lo que sugiere que desempeñan un papel vital en este proceso (Datos complementarios 2).
Prueba de precisión para CircCode
Para investigar la precisión de CircCode, se utilizaron secuencias de prueba generadas por GenRGenS, que utiliza el modelo oculto de Markov para producir secuencias que tienen las mismas características de secuencia (como las frecuencias de diferentes nucleótidos, diferentes codones y diferentes nucleótidos al inicio de la secuencia).
Para este estudio, utilizamos circRNAs traducidos humanos publicados previamente (Yang et al., 2017) como entrada para GenRGenS y generó 10.000 secuencias para probar el código Circum. Repetimos la prueba 10 veces, y en promedio, se predijeron 27 circRNAs traducidos cada vez. El valor FDR se calculó como 0,0027, que es mucho menor que 0,05, lo que indica que los resultados previstos son creíbles.
Además, comparamos los circRNAs traducidos de seres humanos identificados por el CircCode con datos de circRNA asociados a polisomas verificados (Yang et al., 2017). Entre ellos, el 60% de los CIRRNAS fueron identificados por el código CIRCC (Datos complementarios 3).
Influencia de la Profundidad de Secuenciación de Datos Ribo-seq
Para investigar el impacto de la profundidad de secuenciación de datos Ribo-seq en los resultados de identificación de circódigos, primero probamos el efecto de la profundidad de secuenciación en el número de circRNAs traducidos (Figura 2A). Cuando la profundidad de secuenciación era baja, el número predicho de circRNAs traducidos era bajo, y el número de circRNAs traducidos aumentaba con el aumento de la profundidad de secuenciación. El número de ARN traducidos se estabilizó cuando la profundidad de secuenciación alcanzó una cobertura de transcripción lineal de no menos de 10×.FIGURA

Figura 2 (A) Efecto de la profundidad de secuenciación de datos Ribo-seq en el número previsto de circRNAs traducidos. B) El efecto del número de lectura de unión (JRN) en la sensibilidad de los circódigos a diferentes profundidades de secuenciación.
En segundo lugar, también se evaluó la influencia del NMJ en la sensibilidad a diferentes profundidades de secuenciación (Figura 2B). Los resultados mostraron que el NMJ tenía menos impacto en la sensibilidad a medida que aumentaba la profundidad de secuenciación. CircCode también tuvo una mayor sensibilidad al usar datos Ribo-seq con mayor profundidad de secuenciación.
Comparación de CircCode Con Otras Herramientas
Para comparar CircCode con otras herramientas, como CircPro, se utilizó el mismo conjunto de datos Ribo-seq (SRR3495999) de A. thaliana para identificar circRNAs traducidos utilizando seis procesadores, con 16 gigabytes de RAM. CircPro identificó 44 círculos traducidos en 13 minutos, mientras que CircCode identificó 76 círculos traducidos en 20 minutos. Por lo tanto, CircCode es más sensible que CircPro en el mismo nivel de hardware de la computadora, pero lleva más tiempo. CircPro es conciso y consume menos tiempo que CircCode, pero CircCode puede identificar más circRNAs con capacidad de codificación que CircPro.
Conclusiones
Los circRNAs desempeñan un papel importante en la biología, y es crucial identificar con precisión los circRNAs con capacidad de codificación para la investigación posterior. Basado en Python 3, desarrollamos CircCode, una herramienta de línea de comandos fácil de usar que tiene una alta sensibilidad para identificar circRNAs traducidos de lecturas Ribo-Seq con alta precisión. CircCode muestra un buen rendimiento tanto en plantas como en animales. El trabajo futuro agregará el análisis de caracteres descendentes al código de circunferencias al visualizar cada paso del proceso y optimizar la precisión de la predicción.
Disponibilidad y requisitos
CircCode está disponible en https://github.com/PSSUN/CircCode; sistema(s) operativo (s): Linux, lenguajes de programación: Python 3 y R; otros requisitos: bedtools (versión 2.20.0 o posterior), bowtie, STAR, paquetes Python 3 (Biopython, Pandas, rpy2), paquetes R (BASiNET, Bioestrings). Los paquetes de instalación para todo el software requerido están disponibles en la página de inicio de CircCode. Los usuarios no necesitan descargarlos individualmente. La página de inicio de CircCode también proporciona manuales de usuario detallados para referencia. La herramienta está disponible gratuitamente. No hay restricciones de uso para personas no académicas.
Declaración de disponibilidad de datos
Todos los datos pertinentes se encuentran en el manuscrito y en sus archivos de información de apoyo.
Contribuciones de autor
Conceptualización: PS, GL. Curación de datos: PS, GL. Análisis formal: PS, GL. Redacción-Borrador original: PS, GL. Redacción-Revisión y edición: PS, GL.
Financiación
Este trabajo fue apoyado por subvenciones de la Fundación Nacional de Ciencias Naturales de China (subvenciones nos. 31770333, 31370329 y 11631012), el Programa para Talentos Excelentes del Nuevo Siglo en la Universidad (NCET-12-0896) y los Fondos de Investigación Fundamental para las Universidades Centrales (no. GK201403004). Las agencias de financiamiento no tuvieron ningún papel en el estudio, su diseño, la recolección y análisis de datos, la decisión de publicar o la preparación del manuscrito. Los financiadores no desempeñaron ningún papel en el diseño del estudio, la recolección y el análisis de datos, la decisión de publicar o la preparación del manuscrito.
Conflicto de intereses
Los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un conflicto de intereses potencial.
Material suplementario
El Material Suplementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
Datos suplementarios 1 / La secuencia del circRNA traducido predicho y del péptido corto.
Datos complementarios 2 / Resultados de enriquecimiento en GO y enriquecimiento en KEGG para seres humanos y Arabidopsis thaliana.
Datos suplementarios 3 / Comparación de CIRRNAS traducidos predichos con CIRRNAS traducidos validados.
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmatic: un recortador flexible para datos de secuencia illumina. Bioinformática 30, 2114-2120. doi: 10.1093 / bioinformática / btu170
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Brar, G. A., Weissman, J. S. (2015). El perfil de ribosomas revela el qué, cuándo, dónde y cómo de la síntesis de proteínas. NAT. Reverendo Mol. Cell Biol. 16, 651–664. doi: 10.1038 / nrm4069
Resumen de PubMed / Texto completo de CrossRef | Google Scholar
Chen, L.-L., Yang, L. (2015). Regulación de la biogénesis del circRNA. ARN Biol. 12, 381–388. doi: 10.1080/15476286.2015.1020271
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
Chu, Q., Zhang, X., Zhu, X., Liu, C., Mao, L., Ye, C., et al. (2017). Plantcirbase: una base de datos para ARN circulares de plantas. Mol. Planta 10, 1126-1128. doi: 10.1016 / j.molp.2017.03.003
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: alineador universal ARN-seq ultrarrápido. Bioinformática 29, 15-21. doi: 10.1093 / bioinformática / bts635
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Dong, R., Ma, X.-K., Chen, L.-L., Yang, L. (2019). «Genome-wide annotation of circRNAs and their alternative back-splicing / splicing with CIRCexplorer Pipeline», en Epitranscriptomics. Eréctil. Wajapeyee, N., Gupta, R. (Nueva York, NY: Springer New York), 137-149. doi: 10.1007/978-1-4939-8808-2_10
Texto completo de CrossRef | Google Scholar
Dong, R., Ma, X.-K., Li, G.-W., Yang, L. (2018). Circipedia v2: una base de datos actualizada para la anotación circular completa de ARN y la comparación de expresiones. Genómica Proteómica Bioinf. 16, 226–233. doi: 10.1016 / j. gpb.2018.08.001
Texto completo de CrossRef / Google Scholar
Gao, Y., Wang, J., Zhao, F. (2015). CIRI: un algoritmo eficiente e imparcial para la identificación de nuevo ARN circular. Genome Biol. 16, 4. doi: 10.1186 / s13059-014-0571-3
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
Guttman, M., Russell, P., Ingolia, N. T., Weissman, J. S., Lander, E. S. (2013). El perfil de ribosomas proporciona evidencia de que los ARN grandes no codificantes no codifican proteínas. Celda 154, 240-251. doi: 10.1016 / j.cell.2013.06.009
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Hsu, P. Y., Benfey, P. N. (2018). Pequeño pero poderoso: péptidos funcionales codificados por pequeños ORF en plantas. PROTEÓMICA 18, 1700038. doi: 10.1002 / pmic.201700038
Texto completo de CrossRef / Google Scholar
Hsu, P. Y., Calviello, L., Wu, H.-Y. L., Li, F.-W., Rothfels, C. J., Ohler, U., et al. (2016). El perfilado de ribosomas de superresolución revela eventos de traducción no anotados en Arabidopsis. Proc. Natl. Acad. Sci. 113, E7126–E7135. doi: 10.1073 / pnas.1614788113
Texto completo de CrossRef / Google Scholar
Ingolia, N. T., Lareau, L. F., Weissman, J. S. (2011). El perfil de ribosomas de células madre embrionarias de ratón revela la complejidad y la dinámica de los proteomas de mamíferos. Celda 147, 789-802. doi: 10.1016 / j.cell.2011.10.002
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
., E. A., Katahira, I., Vicente, F. F., da, R., Pereira, L. F. P., Lopes, F. M. (2018). BASiNET-BiologicAl Sequences NETwork: a case study on coding and non-coding RNAs identification. Ácidos nucleicos Res. 46, e96–e96. doi: 10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093 / bioinformática / bty948
Texto completo Cruzado / Google Scholar
Kearse, M. G., Wilusz, J. E. (2017). Traducción sin AGOSTO: un nuevo comienzo para la síntesis de proteínas en eucariotas. Genes Dev. 31, 1717–1731. doi: 10.1101 / gad.305250.117
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Kong, L., Zhang, Y., Ye, Z.-Q., Liu, X.-Q., Zhao, S.-Q., Wei, L., et al. (2007). CPC: evaluar el potencial de codificación de proteínas de las transcripciones utilizando características de secuencia y máquina vectorial de soporte. Ácidos nucleicos Res. 35, W345-W349. doi: 10.1093/nar / gkm391
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Langmead, B., Salzberg, S.L. (2012). Alineación rápida de lectura por huecos con pajarita 2. NAT. Methods 9, 357-359. doi: 10.1038 / nmeth.1923
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Meng, X., Chen, Q., Zhang, P., Chen, M. (2017). CircPro: una herramienta integrada para la identificación de circRNAs con potencial codificador de proteínas. Bioinformatics 33, 3314-3316. doi: 10.1093/bioinformática / btx446
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Michel, A. M., Baranov, P. V. (2013). Perfil de ribosomas: un monitor de alta definición para la síntesis de proteínas a gran escala del genoma: perfil de ribosomas. Wiley Interdiscip. Rev. ARN 4, 473-490. doi: 10.1002 / wrna.1172
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Pamudurti, N. R., Bartok, O., Jens, M., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., et al. (2017). Traducción de CircRNAs. Mol. Celda 66, 9-21.e7. doi: 10.1016 / j. molcel.2017.02.021
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Ponty, Y., Termier, M., Denise, A. (2006). GenRGenS: software para generar secuencias y estructuras genómicas aleatorias. Bioinformatics 22, 1534-1535. doi: 10.1093 / bioinformática / btl113
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Rho, M., Tang, H., Ye, Y. (2010). FragGeneScan: predicción de genes en lecturas cortas y propensas a errores. Ácidos nucleicos Res. 38, e191-e191. doi: 10.1093/nar | gkq747
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Slavoff, S.A., Mitchell, A. J., Schwaid, A. G., Cabili, M. N., Ma, J., Levin, J. Z., et al. (2013). Descubrimiento peptídico de péptidos codificados con marco de lectura abierta corta en células humanas. NAT. Chem. Biol. 9, 59–64. doi: 10.1038 / nchembio.1120
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Spealman, P., Naik, A. W., May, G. E., Kuersten, S., Freeberg, L., Murphy, R. F., et al. (2018). UORF conservados no AUG revelados por un nuevo análisis de regresión de datos de perfiles de ribosomas. Genome Res. 28, 214-222. doi: 10.1101 / gr.221507.117
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Vicens, Q., Westhof, E. (2014). Biogénesis de ARN circulares. Celda 159, 13-14. doi: 10.1016 / j.cell.2014.09.005
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Willems, P., Ndah, E., Jonckheere, V., Stael, S., Sticker, A., Martens, L., et al. (2017). La proteómica N-terminal ayudó a perfilar el paisaje inexplorado de iniciación a la traducción en Arabidopsis thaliana. Mol. Celular. Proteomics 16, 1064-1080. doi: 10.1074 / mcp.M116. 066662
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
Wu, J., Mao, X., Cai, T., Luo, J., Wei, L. (2006). Servidor KOBAS: una plataforma basada en la web para la anotación automatizada y la identificación de vías. Ácidos nucleicos Res. 34, W720-W724. doi: 10.1093/nar / gkl167
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
Yang, L., Fu, J., Zhou, Y. (2018). ARN Circulares y Sus Funciones Emergentes en la Regulación Inmunitaria. Delantero. Inmunol. 9, 2977. doi: 10.3389 / fimmu.2018.02977
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Yang, Y., Fan, X., Mao, M., Song, X., Wu, P., Zhang, Y., et al. (2017). Traducción extensa de ARN circulares impulsados por N6-metiladenosina. Cell Res. 27, 626-641. doi: 10.1038 / cr.2017.31
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
Yu, G., Wang, L.-G., Han, Y., He, Q.-Y. (2012). Generador de clústeres: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar