- Introducción
- Estructura y Regulación de ChREBP A través de los Dominios LID/Grace
- Activación de ChREBP por Metabolitos de glucosa
- Cofactores y socios de ChREBP
- El papel de la ChREBP en el Metabolismo de los Carbohidratos y la Producción de Hepatocinas
- La ChREBP como Regulador de la Síntesis de Ácidos Grasos Hepáticos y la Secreción de VLDL
- Regulación del Metabolismo de la fructosa por ChREBP en Hígado e Intestino
- Reglamento de la BDK:Eje PPM1K en el hígado
- Se requiere ChREBP para la Regulación Mediada por Glucosa de FGF21
- Papel de la ChREBP en la Red Interorgánica Que Controla la Homeostasis Energética
- Papel de la ChREBP hepática en el Control del Equilibrio de Sensibilidad a la Insulina
- La ChREBP adiposa Vincula la lipogénesis con la Sensibilidad a la insulina
- La nueva interacción entre la Lipasa Sensible a hormonas y la ChREBP en el tejido Adiposo
- Conclusión y Direcciones futuras
- Contribuciones de los autores
- Financiación
- Declaración de Conflicto de Intereses
Introducción
El aumento del consumo de azúcares simples como sacarosa y jarabe de maíz de alta fructosa en los últimos años ha llevado a un mayor riesgo de enfermedades metabólicas como obesidad, dislipidemia, diabetes tipo 2 y/o enfermedad hepática grasa no alcohólica (NAFLD). El hígado es el principal órgano responsable de la conversión del exceso de carbohidratos dietéticos en grasa. Los triglicéridos (TG) resultantes se pueden empaquetar en lipoproteínas de muy baja densidad (VLDL) y secretarse en la circulación, almacenarse como gotitas de lípidos o metabolizarse a través de la vía de oxidación beta. La insulina secretada en respuesta al aumento de la glucosa en sangre, estimula la expresión de genes de la síntesis de ácidos grasos de novo (lipogénesis) a través de la proteína de unión al elemento regulador esterol del factor de transcripción-1c (SREBP-1c) (Foretz et al., 1999). El SREBP-1c actúa en sinergia con otro factor de transcripción llamado proteína de unión al elemento de respuesta de carbohidratos (ChREBP), que media la respuesta a los carbohidratos dietéticos. La estructura de la proteína ChREBP contiene un dominio inhibitorio de glucosa bajo (LID) y un elemento conservado de activación de la respuesta de glucosa (GRACE) ubicado en su extremo N (Li et al., 2006). La activación del dominio GRACE por los metabolitos de glucosa promueve la actividad transcripcional de ChREBP y la unión a una secuencia altamente conservada llamada elemento de respuesta de carbohidratos (tarea). La tarea está presente en los promotores de los genes diana de ChREBP, que codifican enzimas clave de la lipogénesis de novo, incluida la L-piruvato quinasa (L-pk), una enzima limitadora de velocidad en la glucólisis, la sintasa de ácidos grasos (Fas), la acetil-COA carboxilasa (Acc) y la estearoil-CoA desaturasa (Scd1) (Kawaguchi et al., 2001). Un estudio reciente informó de la interdependencia entre ChREBP (activado por glucosa) y SREBP-1c (activado por insulina) para la inducción completa de la expresión génica glucolítica y lipogénica en el hígado (Linden et al., 2018). El restablecimiento viral de la forma nuclear activa de SREBP-1c en el hígado de ratones con deficiencia de ChREBP (ChREBPKO) normalizó la expresión génica lipogénica, sin tener ningún efecto en el rescate de la expresión génica glucolítica. El experimento mirror, en el que se indujo la expresión de ChREBP en el hígado de ratones knockout SREBP-1c, rescató la expresión génica glucolítica, pero sorprendentemente no la expresión génica lipogénica, a pesar del papel bien conocido de ChREBP en el control de los genes de síntesis de ácidos grasos. Sin embargo, este estudio sugiere la importancia de la doble acción de ChREBP y SREBP-1c en los genes de control involucrados en la regulación de la síntesis de ácidos grasos (Linden et al., 2018).
El ChREBP está altamente enriquecido en el hígado y se ha estudiado como regulador maestro del metabolismo lipídico (Iizuka et al., 2004; Osorio et al., 2016). La ChREBP también se expresa significativamente en los islotes pancreáticos, el intestino delgado, el músculo esquelético y, en menor medida, en el riñón y el cerebro (ver Richards et al., 2017 para revisión). Curiosamente, otra isoforma de ChREBP, ChREBPß, que se origina a partir de un primer promotor de exones alternativo, se identificó por primera vez en el tejido adiposo (Herman et al., 2012) y más tarde descrito en otros tipos de células (ver Abdul-Wahed et al., 2017 para revisión). Como discutiremos, ChREBPß se describe como una isoforma constitutivamente activa. Se espera que el trabajo futuro aborde las funciones respectivas del ChREBP y el ChREBPß en la regulación del metabolismo de la glucosa y los lípidos, así como identificar sus objetivos específicos o superpuestos.
Estructura y Regulación de ChREBP A través de los Dominios LID/Grace
ChREBP pertenece a la familia Mondo de factores de transcripción bHLH/Zip. El dominio N-terminal (residuos 1-251) contiene dos señales de exportación nuclear (NES) y una señal de localización nuclear (NLS) que regulan la localización subcelular al interactuar con el mantenimiento de la región cromosómica 1 (CRM1), también conocidas como proteínas exportina 1 y/o 14-3-3 (Sakiyama et al., 2008). La región C-terminal contiene un dominio de poliprolina, un dominio bHLH / LZ (660-737 residuos) y un dominio tipo cremallera de leucina (residuos tipo cremallera, 807-847) que están asociados con cofactores y unión al ADN (Yamashita et al., 2001; Fukasawa et al., 2010; Ge et al., 2012). La localización y la activación transcripcional del ChREBP están determinadas por la disponibilidad de nutrientes. La regulación de la ChREBP mediada por glucosa ocurre principalmente a nivel del módulo de detección de glucosa (GSM) o región conservada mondo (MCR), que está compuesta por los dominios LID y GRACE, como se menciona en la introducción (Figura 1A; Li et al., 2006; Singh e Irwin, 2016). En 2012, Herman et al. (2012) describieron otra isoforma de ChREBP, ChREBPß, que se transcribe de un primer promotor de exón alternativo 1b al exón 2 (Figura 1B). Esta transcripción se traduce del exón 4, generando una proteína más corta de 687 aminoácidos (la isoforma ChREBP de longitud completa, rebautizada α, contiene 864 aminoácidos, llamados ChREBP en el manuscrito) en la que faltan los dos NES, NLS y el dominio LID. ChREBPß es altamente activo en el tejido adiposo blanco de una manera dependiente de GLUT-4 y se sugiere que está regulado directamente por ChREBPa ya que se identificó una secuencia de tareas en el promotor de exones 1β (Herman et al., 2012; gráfico 1B). La regulación de ChREBPß por ChREBPa sugiere la existencia de un bucle de alimentación hacia adelante que potencialmente exacerba la respuesta a la glucosa en condiciones hiperglucémicas. Sin embargo, es necesario aclarar el mecanismo o mecanismos reguladores de la isoforma ChREBPß y, lo que es más importante, su función específica.
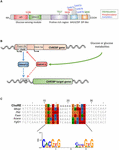
Figura 1. A) Estructura de la proteína de unión al elemento de respuesta a los carbohidratos α (ChREBPa). ChREBPa se compone de 864 aminoácidos y contiene varios dominios reguladores. En el extremo N, la proteína contiene un módulo de detección de glucosa compuesto por el dominio inhibitorio de glucosa baja (LID) y el elemento conservado activado por glucosa (GRACE). La proteína también contiene un dominio rico en poliprolina, un bHLH/LZ y un dominio tipo cremallera de leucina ubicado en el terminal C. Las modificaciones postraduccionales se indican en sus respectivos residuos, fosforilación (rojo), acetilación (azul) y las O-Glcnacilaciones recientemente identificadas (verde). B) Estructura génica del gen ChREBP y generación de las dos isoformas de ChREBP α y β. El ChREBPß se transcribe de un primer promotor de exón alternativo 1b. Esta transcripción se traduce del exón 4 generando una proteína más corta de 687 aminoácidos en la que faltan los dos NES, el NLS y el dominio del párpado. Se ha sugerido que la isoforma ChREBPß está regulada directamente por ChREBPa ya que se identificó una secuencia de tareas en el promotor de exones 1b. Actualmente se desconoce si ambas isoformas ChREBP α y β se unen a la tarea. Figura adaptada de Herman et al. (2012). (C) Alineación múltiple de secuencias de consenso de tareas presentes en varios promotores de genes diana de ChREBP. La alineación basada en nucleótidos se presenta en la parte superior de la figura junto con la tarea de secuencia de consenso descrita en Poungvarin et al. (2015). El logotipo correspondiente a la secuencia de consenso asociada a esta alineación en particular también está representado.
Activación de ChREBP por Metabolitos de glucosa
En condiciones de ayuno, la activación dependiente del glucagón de la proteína quinasa A (PKA) (Kawaguchi et al., 2002) fosforila ChREBP en residuos Ser196 y Thr666, lo que conduce a la unión de ChREBP a la proteína 14-3-3 y su retención en el citosol (Kawaguchi et al., 2001, 2002; Davies et al., 2008). La proteína quinasa activada por AMP (AMPK), un sensor central de energía celular, también fosforila la ChREBP en el residuo Ser568, lo que a su vez disminuye la unión de la ChREBP a los promotores de sus genes diana (Kawaguchi et al., 2002; Sato et al., 2016). Se demostró que los metabolitos generados durante el ayuno, como los cuerpos AMP y cetónicos producidos a partir de la oxidación de ácidos grasos, desempeñan un papel inhibidor alostérico al alterar la afinidad de la proteína ChREBP y 14-3-3, mejorando la estabilización compleja y favoreciendo la retención citosólica (Sakiyama et al., 2008; Nakagawa et al., 2013; Sato et al., 2016). En respuesta a los carbohidratos, el ChREBP se regula en los niveles transcripcional, traslacional y post-traslacional. El aumento de las concentraciones de glucosa después de una comida promueve la síntesis de metabolitos intermedios como la xilulosa-5-fosfato (X5P), propuesta inicialmente como activador de la proteína fosfatasa 2A (PP2A) (Kawaguchi et al., 2001; Kabashima et al., 2003). El PP2A fue previamente descrito para desfosforilar ChREBP en el residuo Ser196 permitiendo su translocación al núcleo donde es desfosforilado de una manera dependiente de X5P y PP2A (en Thr666 y Ser568). Sin embargo, este modelo fue cuestionado a lo largo de los años y se propuso que otros metabolitos como la glucosa-6-fosfato (G6P) fueran activadores potenciales de la translocación/actividad de ChREBP (Dentin et al., 2012). McFerrin et al. (2012) identificaron un motivo putativo para la unión G6P (253-SDTLFT-258) en el dominio GRACE, que también se conserva en MondoA, un ortólogo ChREBP/MondoB (ver Richards et al., 2017 para revisión). De acuerdo con esta hipótesis, el G6P podría promover un cambio de conformación alostérica que induce una conformación abierta para el ChREBP, facilitando la interacción con cofactores y la posterior translocación al núcleo (McFerrin et al., 2012).
Dentro del núcleo, la ChREBP se puede modificar a través de la O-GlcNAcilación, una modificación postraduccional dependiente del metabolismo de la glucosa, e identificada como importante para la actividad transcripcional de la ChREBP (Guinez et al., 2010). La O-GlcNAcilación ocurre en residuos de serina y treonina a través de la actividad de la O-GlcNAc transferasa (OGT), una enzima que agrega residuos de N-acetilglucosamina (GlcNAc) a las proteínas diana, modificando así su actividad, estabilidad y/o ubicación subcelular. Yang et al. (2017) revelaron recientemente varios residuos de ChREBP modificados por O-GlcNacilación. Las mutaciones de estos residuos en los dominios bHLH / ZIP y la dimerización y el dominio de localización citoplasmática (DCD) han permitido la identificación de Thr517 y Ser839 como sitios esenciales para la activación dependiente de la glucosa del ChREBP (Figura 1A). La ChREBP también se puede modificar por acetilación a través de la actividad de la histona acetiltransferasa de p300 (Bricambert et al., 2010). El acetilado p300 activado por glucosa crea ChREBP en Lys672 y aumenta su actividad transcripcional al mejorar su reclutamiento en la secuencia de tareas, cuya secuencia de unión de consenso óptima es CAYGYCnnnnnCRCRTG (Figura 1C). Poungvarin et al. (2015) analizaron los sitios de unión de ChREBP por ChIP-seq en el hígado y el tejido adiposo blanco de ratones alimentados con una dieta alta en carbohidratos y libre de grasas. Informaron que la unión de la ChREBP se enriquece en las vías involucradas en la señalización de insulina, las uniones adherentes y el cáncer, lo que sugiere una nueva participación de la ChREBP en la génesis tumoral y la progresión del cáncer. Además, un estudio reciente informó la importancia de la ChREBP en el carcinoma hepatocelular (CHC) (Ribback et al., 2017). Los autores encontraron que la deleción genética de ChREBP (en ratones ChREBPKO) afectó la hepatocarcinogénesis impulsada por la sobreexpresión de la proteína quinasa B/Akt en ratones. Además, la inhibición de la ChREBP mediada por siRNA en células HCC de ratón y/o humanos dio lugar a una disminución de la proliferación y la apoptosis.
Cofactores y socios de ChREBP
En los últimos años se identificaron varios cofactores y/o socios de ChREBP (ver Richards et al., 2017 para revisión). Max like protein x (Mlx), un factor de transcripción bHLH/LZ, fue el primero identificado como un socio de unión común de la familia Mondo (Stoeckman et al., 2004). La dimerización de ChREBP con Mlx es necesaria tanto para la translocación nuclear en respuesta a la glucosa como para la unión a elementos de tareas. Receptores nucleares como el factor nuclear de hepatocitos 4α (HNF4a) y el receptor farnesoide X (FXR) también se describieron como socios de ChREBP. HNF4a interactúa físicamente con ChREBP al unirse a la región de repetición directa-1 (DR-1) en el promotor de los genes diana de ChREBP (Adamson et al., 2006; Meng et al., 2016). Además, se demostró que las proteínas coactivadoras transcripcionales p300 / CBP estabilizan el complejo ChREBP / HNF4a (Burke et al., 2009). Las proteínas coactivadoras transcripcionales p300 / CBP desempeñan un papel central en la coordinación e integración de múltiples eventos dependientes de la señal con el aparato de transcripción. Otra propiedad clave de p300 / CBP es la presencia de la actividad de la histona acetiltransferasa (HAT), que dota a p300/CBP de la capacidad de influir en la actividad de la cromatina modulando las histonas nucleosómicas. En los hepatocitos humanos, la unión de FXR al complejo ChREBP-HNF4a desencadena la liberación de ChREBP desde CBP/p300, lo que lleva al reclutamiento de la histona desacetilasa SMRT en el promotor Lpk, actuando así como copresor de la actividad transcripcional de ChREBP (Caron et al., 2013). Además, la actividad de CBP / p300 HAT modifica la ChREBP en Lys 672, lo que lleva a su activación transcripcional en respuesta a la glucosa (Bricambert et al., 2010).
Bricambert et al. (2018) identificaron recientemente el homeodominio de la planta de histona desmetilasa finger 2 (Phf2), que pertenece a la familia de la histona lisina desmetilasa (KDM7), como un nuevo cofactor de ChREBP. La interacción entre Phf2 y ChREBP mejora la activación transcripcional de ChREBP al borrar las marcas de metilo H3K9 en el promotor de sus genes diana. Curiosamente, el reclutamiento conjunto específico de Phf2 y ChREBP al promotor del factor nuclear eritroide 2 como 2 (Nrf2) contribuye al efecto protector de Phf2 contra el aumento de especies reactivas de oxígeno (ROS) y la progresión de NAFLD en el contexto de la hiperglucemia (Bricambert et al., 2018).
El papel de la ChREBP en el Metabolismo de los Carbohidratos y la Producción de Hepatocinas
La ChREBP como Regulador de la Síntesis de Ácidos Grasos Hepáticos y la Secreción de VLDL
La enfermedad hepática grasa no alcohólica es un sello distintivo del síndrome metabólico, y los estudios en seres humanos revelan que la lipogénesis de novo contribuye a aproximadamente el 25% de los lípidos hepáticos totales en pacientes con EHGNA (Donnelly et al., 2005). En estados resistentes a la insulina, la hiperglucemia y la hiperinsulinemia mejoran la lipogénesis en parte a través de la activación de ChREBP y SREBP-1c. La inhibición de ChREBP en el hígado de ratones ob/ob obesos y resistentes a la insulina, a través de ARNi o ablación genética, conduce a la reversión de la esteatosis hepática (Dentin et al., 2006; Iizuka et al., 2006). La secreción alterada de VLDL por el hígado también contribuye a la patogénesis de NAFLD. La proteína de transferencia de triglicéridos microsomales (MTTP) es la proteína encargada de ensamblar y secretar las lipoproteínas que contienen apolipoproteína B. La deficiencia de MTTP en ratones y humanos causa hipolipidemia e hígado graso. La regulación de esta proteína se ha asociado con unos pocos elementos cis altamente conservados en su promotor, incluidos los dominios reguladores críticos positivos y negativos (Cuchel et al., 2013; Hussain et al., 2011). Recientemente, la ChREBP se señaló como un regulador potencial de MTTP, ya que la falta de ChREBP funcional en el hígado suprime la expresión de Mttp y el ensamblaje y secreción de VLDL (Niwa et al., 2018). Sin embargo, dado que no se pudo identificar claramente ninguna tarea en el promotor de Mttp, será necesario un análisis más detallado para identificar el mecanismo con el que ChREBP regula el Mttp.
Regulación del Metabolismo de la fructosa por ChREBP en Hígado e Intestino
El vínculo entre ChREBP y el metabolismo de la fructosa se evidenció por primera vez mediante el análisis fenotípico de ratones knockout de ChREBP (ratones ChREBPKO). Se informó que los ratones ChREBPKO morían a los pocos días de alimentarse con dieta alta en fructosa (HFrD) (Iizuka et al., 2004). Esta intolerancia importante a la fructosa se atribuyó a la reducción en la expresión de fructocinasa y triosa quinasa, dos enzimas necesarias para el metabolismo de la fructosa (Iizuka et al., 2004). Kim et al. (2016) informaron más tarde sobre la importancia de la ChREBP para la conversión eficiente de fructosa en glucosa en el aclaramiento de fructosa en el hígado y en todo el cuerpo, pero también, bajo la ingestión de fructosa, la ChREBP podría contribuir a la hiperglucemia al transactivar directamente la expresión de G6pc, un gen clave de la gluconeogénesis. Este efecto podría conducir a un círculo vicioso en el que el consumo de fructosa exacerba la producción de glucosa a través de la actividad de ChREBP (Kim et al., 2016). Al año siguiente, el estudio de Zhang et al. (2017) informaron que los ratones ChREBPKO alimentados con HFrD desarrollan lesión hepática grave debido a la sobreactivación del estrés del retículo endoplásmico y la apoptosis de hepatocitos mediada por la proteína homóloga de unión a potenciadores de CCAAT (CHOP). La apoptosis en los hepatocitos de estos ratones se relacionó muy probablemente con un aumento de la biosíntesis del colesterol, ya que la inhibición de esta vía a través de la HMG-COA reductasa (HMGCR) o la inhibición de SREBP2 rescató a los ratones ChREBPKO de una lesión hepática inducida por HFrD. La falta de ChREBP también se asoció recientemente con una desregulación del metabolismo de sacarosa y fructosa que condujo a intolerancia al azúcar y malabsorción en ratones (Kato et al., 2018). Estos efectos se asociaron con una disminución de la expresión de la sacarosa-isomaltasa intestinal (SI), que digiere la sacarosa en glucosa y fructosa, los transportadores de glucosa 5 (Glut5) y 2 (Glut2) y la enzima ketohexocinasa (Khk), que regula la fructólisis (Figura 2). La desregulación de estas enzimas puede conducir a la acumulación de sacarosa y fructosa sin digerir, con repercusiones potenciales en la composición de la microbiota intestinal. La comparación entre ChREBPKO y ratones knockout de ChREBP específicos para el hígado (ChREBPLiverKO) alimentados con HFrD había revelado previamente que la deficiencia hepática de ChREBP por sí sola no conduce a intolerancia a la fructosa, pero que la deficiencia de ChREBP en el intestino delgado es probablemente responsable del deterioro en la tolerancia a la fructosa observado en estos ratones (Kim et al., 2017). En conjunto, estos estudios subrayan la importancia del ChREBP en la regulación del metabolismo de la fructosa y subrayan la necesidad de una mejor comprensión de su papel y regulación en el intestino delgado.
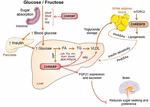
Figura 2. El ChREBP regula múltiples vías de señalización / metabólicas en respuesta a la glucosa y la fructosa. La ChREBP se expresa en varios tejidos, incluidos el intestino, el hígado y el tejido adiposo blanco. En estos tipos celulares, en respuesta a la glucosa y/o fructosa, el ChREBP se activa e induce un programa genético específico como se indica en la figura. En el intestino, se describió la estimulación de la expresión de SI, Glut5, Glut2 y Cetohexocinasa (Khk) por ChREBP (directa o indirectamente) para mejorar la tolerancia a la sacarosa y la absorción de fructosa. En el hígado, la ChREBP es un modulador clave de la expresión génica de la proteína de transferencia de triglicéridos (Mttp) glucolítica, lipogénica y microsomal, controlando así la acumulación de ácidos grasos y la exportación de VLDL desde el hígado. El ChREBP también regula la producción de hepatocinas como el factor de crecimiento de fibroblastos 21 (FGF21). Este eje hígado-cerebro expande la función de la ChREBP hepática de un regulador hepático a un modulador sistémico que afecta no solo el manejo del sustrato en el hígado, sino también la preferencia de nutrientes. La activación de la ChREBP en el tejido adiposo blanco está relacionada con la mejora de la homeostasis metabólica al producir señales circulantes protectoras. Se informó que una nueva clase de lípidos de mamíferos caracterizados por un enlace de éster ramificado entre un ácido graso y un ácido hidroxigraso (ácido palmítico hidroxilesteárico) ejerce efectos beneficiosos sobre la homeostasis de la glucosa a través de la modulación directa e incretina de la función de las células β, la captación de glucosa adiposa mejorada y la reducción de la inflamación. Curiosamente, mTORC2 fue recientemente identificado como un nuevo regulador de la isoforma de ChREBPß en las células adiposas.
Reglamento de la BDK:Eje PPM1K en el hígado
El primer paso comprometido del catabolismo de aminoácidos de cadena ramificada (BCAA) está regulado por el complejo cetoácido deshidrogenasa de cadena ramificada (BCKDH) que está controlado por dos enzimas, la alfa-ceto ácido deshidrogenasa cinasa (BDK) de cadena ramificada y la proteína fosfatasa, Mg2+/Mn22+ dependiente de 1K (PPM1K). White et al. (2018) asociaron recientemente la ChREBP con la regulación ascendente de BDK y la regulación descendente de PPM1K en el hígado e identificaron un motivo de tareas conservadas en el promotor de ambos genes. Se observó una correlación positiva entre la expresión de BDK y otros genes diana típicos de ChREBP (Fasn, Pklr, ChREBPß) en hígados de ratas alimentadas con una dieta alta en glucosa o fructosa. A nivel fisiológico, el aumento de la relación BDK: PPM1K condujo a la fosforilación y activación de la ATP-citrato liasa (ACLY), estimulando así la lipogénesis de novo. Estos hallazgos revelan que BDK y PPM1K pueden ser nuevos genes activadores de la lipogénesis regulados por ChREBPß. Dado su papel en la regulación del metabolismo de lípidos, glucosa y aminoácidos, BDK y PPM1K podrían considerarse como potenciales dianas terapéuticas en el hígado en un futuro próximo (White et al., 2018).
Se requiere ChREBP para la Regulación Mediada por Glucosa de FGF21
ChREBP se asoció recientemente a la producción y secreción de hepatocinas como el factor de crecimiento de fibroblastos 21 (FGF21) (Iizuka et al., 2009; Dushay et al., 2015, Iroz et al., 2017). FGF21 es una hormona metabólica sintetizada por el hígado con múltiples efectos beneficiosos en tejidos periféricos (Kharitonenkov et al., 2005; Badman et al., 2007; Markan et al., 2014). Hasta hace poco, la FGF21 se consideraba una hormona en ayunas que mejora la oxidación de ácidos grasos, la cetogénesis y la lipólisis bajo el control transcripcional del receptor activado por proliferadores de peroxisomas α (PPARa) (Inagaki et al., 2007). Se ha identificado previamente una tarea en el promotor Fgf21 tanto en ratones (-74 a -52 pb) como en humanos (-380 a -366 pb) (Iizuka et al., 2009), pero los estudios funcionales han faltado hasta hace poco. Se informó que el consumo de glucosa y fructosa condujo a una rápida elevación de los niveles de FGF21 en voluntarios sanos y pacientes con síndrome metabólico (Dushay et al., 2015). Estudios adicionales también reportaron un vínculo mecánico entre el FGF21 derivado de ChREBP y la preferencia de macronutrientes a través de un eje hígado-cerebro (Talukdar et al., 2016; von Holstein-Rathlou et al., 2016). Este eje hígado-cerebro expande la función de ChREBP de un regulador metabólico hepático a un modulador sistémico, afectando no solo el manejo del sustrato hepático, sino también el comportamiento de alimentación global(Abdul-Wahed et al., 2017).
Papel de la ChREBP en la Red Interorgánica Que Controla la Homeostasis Energética
Papel de la ChREBP hepática en el Control del Equilibrio de Sensibilidad a la Insulina
Nuestro laboratorio informó previamente que la ChREBP actúa como un modulador clave de la composición de ácidos grasos hepáticos y la sensibilidad a la insulina en el contexto de enfermedades hepáticas no alcohólicas y alcohólicas (ver Abdul-Wahed et al., 2017 para revisión). Los ratones que sobreexpresaron ChREBP desarrollaron mayor esteatosis hepática que los controles, pero curiosamente se mantuvieron libres de complicaciones metabólicas y no desarrollaron resistencia a la insulina. El análisis lipidómico ha revelado que la esteatosis mediada por ChREBP se asocia con una disminución de ácidos grasos saturados y un aumento de ácidos grasos monoinsaturados, estos últimos que se han demostrado que están asociados con efectos beneficiosos mediados por ChREBP sobre la sensibilidad a la insulina (Benhamed et al., 2012). Estos resultados demuestran el papel del ChREBP en la partición lipídica y sugieren que especies lipídicas específicas, cuando están presentes en el lugar y el tiempo adecuados, pueden desencadenar señales que modulan la adaptación al estrés metabólico (Benhamed et al., 2012; Bricambert et al., 2018). Curiosamente, Jois et al. (2017) también sugirieron un papel protector para la ChREBP hepática con respecto a la homeostasis de glucosa en todo el cuerpo y la sensibilidad a la insulina. Los ratones ChREBPLiverKO exhiben una tolerancia a la glucosa empeorada, mientras que están protegidos de la esteatosis hepática. La deleción hepática de ChREBP también produjo cambios en la expresión génica de los tejidos adiposos blancos y marrones, lo que sugiere comunicación entre tejidos. La contribución del ChREBP al equilibrio energético de todo el cuerpo puede, por lo tanto, depender de su regulación de las especies lipídicas y/o la producción de hepatoquinas que contribuyen a la coordinación entre tejidos de la homeostasis energética (Jois et al., 2017).
La ChREBP adiposa Vincula la lipogénesis con la Sensibilidad a la insulina
La señalización deficiente de la insulina en el tejido adiposo es una característica crítica de la resistencia a la insulina. Los estudios han informado que la activación de la ChREBP en el tejido adiposo blanco puede mejorar la homeostasis metabólica al producir señales circulantes protectoras(Yore et al., 2014; Tang et al., 2016). Se informó que una clase de lípidos de mamíferos caracterizados por un enlace de éster ramificado entre un ácido graso y un ácido hidroxigraso, ácido palmítico hidroxilesteárico (PAHSA), ejerce efectos beneficiosos sobre la homeostasis de la glucosa a través de la modulación directa y mediada por la incretina de la función de las células β, la captación de glucosa y la reducción de la inflamación (Yore et al., 2014). Del mismo modo, el knockout de ChREBP adiposo específico (ChREBPadiposeKO), que presenta tasas de lipogénesis bajas en el tejido adiposo, es resistente a la insulina con una acción de insulina alterada en el hígado, los músculos y el tejido adiposo blanco en condiciones de dieta alta en grasas y comida. Los ratones ChREBPadiposeKO tienen niveles séricos más bajos de PAHSA, mientras que la suplementación con PAHSA, en particular el isómero 9-PAHSA, rescata la resistencia a la insulina global de ChREBPadiposeKO y la inflamación del tejido adiposo, lo que confirma que la pérdida de ChREBP adiposo es suficiente para causar resistencia a la insulina (Vijayakumar et al., 2017). Un estudio reciente identificó el objetivo mecánico del complejo 2 de rapamicina (mTORC2) como un nuevo regulador de la ChREBP (especialmente la isoforma β) en las células adiposas. Ablación específica del compañero insensible a la rapamicina de mTOR (Rictor) en adipocitos maduros, alteración de la captación de glucosa estimulada por la insulina en el tejido adiposo que conduce a la regulación descendente del ChREBPß y la expresión génica diana involucrada en el control de la lipogénesis (Tang et al., 2016). De acuerdo con una importante interferencia del hígado adiposo mediada por ChREBP, estos efectos se asocian con resistencia a la insulina hepática y aumento de la gluconeogénesis. En conjunto, estos estudios apoyan un papel importante de la ChREBP adiposa en la activación de señales sensibles a la insulina(Tang et al., 2016).
La nueva interacción entre la Lipasa Sensible a hormonas y la ChREBP en el tejido Adiposo
La ChREBP se identificó recientemente como un socio de la enzima lipolítica lipasa sensible a hormonas (HSL) en el tejido adiposo (Morigny et al., 2019). Se demostró que el derribo de HSL en adipocitos humanos y tejido adiposo de ratón mejora la sensibilidad a la insulina e induce el alargamiento de la enzima de ácidos grasos de cadena muy larga (Elovl6). Elov16 es una enzima microsomal que regula la elongación de los ácidos grasos saturados y monoinsaturados C12-16 de manera dependiente de ChREBP (Morigny et al., 2019). A nivel mecanicista, la interacción física entre HSL y ChREBP afectó la translocación nuclear de ChREBPa y la subsiguiente inducción de ChREBPß y genes diana, en particular Elovl6 (Morigny et al., 2019). Este estudio revela una nueva regulación de la ChREBP en el tejido adiposo. La inhibición de la interacción entre HSL y ChREBP puede conducir a estrategias terapéuticas potenciales para mejorar la sensibilidad a la insulina en las células grasas.
Conclusión y Direcciones futuras
ChREBP es ahora un sensor de carbohidratos bien establecido. Aunque la mayoría de los estudios se han dedicado a su implicación en el control de las vías glucolíticas y lipogénicas, los datos recientes también han revelado nuevas contribuciones de la ChREBP en hepatocitos y en células grasas, donde podría ser instrumental en la producción de hepatocinas y/o lipoquinas que desencadenan la interferencia entre órganos. Como se ha comentado, los cofactores (modificadores epigenéticos) y/o parejas (LSH adiposa) recientemente identificados en estos tejidos también pueden representar estrategias terapéuticas potenciales para el HGNA y/o para mejorar la sensibilidad sistémica a la insulina. Estudios recientes también han apoyado la importancia de la ChREBP en la regulación del metabolismo de la fructosa y han subrayado la necesidad de una mejor comprensión de su papel y regulación en el intestino delgado. Por último, la identificación de objetivos específicos y/o superpuestos de ChREBPa y ChREBPß en tipos celulares clave, así como la determinación de su impacto específico en la sensibilidad a la insulina, serán de especial importancia en los próximos años.
Contribuciones de los autores
Todos los autores enumerados han hecho una contribución sustancial, directa e intelectual al trabajo y lo han aprobado para su publicación.
Financiación
El laboratorio de Postic (U1016-Institut Cochin) está apoyado por subvenciones de la Red ChroME (Marie Curie Skłodowska Action H2020-MSCA-ITN-2015-675610), la Fundación para la Investigación Médica (FRM) (DEQ20150331744) y la ANR-15-CE14-0026-Hepatoquindo.
Declaración de Conflicto de Intereses
Los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un conflicto de intereses potencial.