- Introducción
- Las funciones múltiples de la quimasa
- Quimasa en mastocitos
- Múltiples funciones enzimáticas de la quimasa
- La función enzimática de la quimasa en NASH
- Participación de la quimasa en Modelos animales de NASH
- Efecto del Inhibidor de la Quimasa en Modelos Animales de NASH
- Efecto del Inhibidor de la Quimasa en Modelos Animales de NASH
- El mecanismo de Inflamación Hepática Atenuado por el inhibidor de la quimasa
- Mecanismo de la esteatosis hepática Atenuado por el Inhibidor de la quimasa
- El mecanismo de Fibrosis Hepática Atenuado por el Inhibidor de la quimasa
- Conclusión
- Contribuciones de autor
- Declaración de Conflicto de Intereses
Introducción
La enfermedad hepática grasa no alcohólica (NAFLD) ha sido reconocida como la forma más común de enfermedad hepática (Angulo, 2002; Clark et al., 2002). La esteatohepatitis no alcohólica (EHNA) imita la hepatitis alcohólica a pesar de la ausencia de antecedentes de consumo de alcohol (Ludwig et al., 1980). La EHGNA y la EHNA se asocian con el síndrome metabólico resultante de la obesidad, la resistencia a la insulina, la hiperlipidemia y la hipertensión. La EHGNA se considera la enfermedad hepática más común y se presenta típicamente como esteatosis hepática simple (Tiniakos et al., 2010). Por el contrario, la EHNA se caracteriza por esteatosis grave, inflamación lobular y fibrosis hepática (Powell et al., 1990; Bertot y Adams, 2016). Aunque el mecanismo responsable del desarrollo de NASH sigue sin estar claro, se propone que la NASH sea causada por un proceso de «impacto múltiple», con la esteatosis hepática como el «primer impacto» y los impactos posteriores, como la inflamación, el estrés oxidativo y las endotoxinas (Tilg y Moschen, 2010). La EHNA está estrechamente relacionada con el síndrome metabólico, y varios estudios clínicos han investigado el tratamiento terapéutico de la EHNA centrándose en los síntomas de la diabetes, la hiperlipidemia y la hipertensión (Georgescu et al., 2009; Park et al., 2010; Mahady et al., 2011). Sin embargo, no se han establecido agentes terapéuticos comúnmente aceptados.
La quimasa puede estar implicada en la patogénesis de la fibrosis hepática. La actividad de la quimasa aumentó significativamente en el hígado de pacientes con fibrosis o cirrosis y hubo una correlación significativa entre el nivel de quimasa y el grado de fibrosis (Komeda et al., 2008). Aunque no se ha notificado un aumento de la actividad de la quimasa en pacientes con NASH, se ha observado en modelos animales de NASH (Tashiro et al., 2010; Masubuchi et al., 2013; Miyaoka et al., 2017). Por el contrario, la inhibición de la quimasa mediante el uso de inhibidores de moléculas bajas resultó en una reducción significativa de la inflamación, la esteatosis y la fibrosis en modelos de NASH de ratas y hámsteres (Tashiro et al., 2010; Masubuchi et al., 2013; Miyaoka et al., 2017). Estos hallazgos indican que la quimasa puede estar implicada en la inflamación, la esteatosis y la fibrosis durante el desarrollo y la progresión de la EHNA (Figura 1).
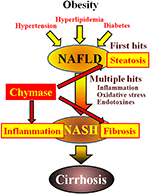
FIGURA 1. La EHGNA y la EHNA están vinculadas al síndrome metabólico por la obesidad, la resistencia a la insulina, la hiperlipidemia y la hipertensión. Se cree que el NASH se desarrolla a través de un proceso de «golpe múltiple», con esteatosis hepática como el «primer golpe» y golpes posteriores, como inflamación, estrés oxidativo y endotoxinas, y se caracteriza por esteatosis severa, inflamación y fibrosis. La quimasa puede estar involucrada en la progresión de la esteatosis, la inflamación y la fibrosis en el hígado.
Las funciones múltiples de la quimasa
Quimasa en mastocitos
La quimasa (EC 3.4.21.39) se expresa en los gránulos secretores de los mastocitos. La quimasa se produce como proquimasa inactiva dentro de gránulos secretores, y requiere dipeptidil peptidasa I (DPPI) para su activación. DPPI es una proteinasa tiólica y su pH óptimo es de 6,0. El valor de pH óptimo es consistente con la función propuesta de DPPI para activar proquimasa, ya que el pH dentro de los gránulos secretores se regula a un pH de 5,5 (De Young et al., 1987) (gráfico 2). Sin embargo, la quimasa no tiene actividad enzimática dentro de los mastocitos a este pH, porque el pH óptimo para la quimasa está entre 7 y 9 (Takai et al., 1996, 1997). Después de la activación de los gránulos de mastocitos por estímulos como inflamación y lesión, la quimasa se libera y exhibe una función enzimática a su pH óptimo 7,4 (Figura 2).
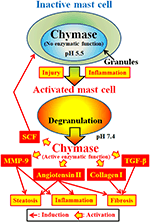
FIGURA 2. La quimasa se almacena en los gránulos secretores de los mastocitos inactivos. El pH dentro de los gránulos se mantiene a un pH de 5,5, una condición en la que la quimasa no tiene actividad enzimática. La quimasa exhibe sus funciones enzimáticas, como la formación de angiotensina II, MMP-9, TGF-β, colágeno I y SCF, al liberarse de los gránulos de mastocitos, tras la activación por inflamación y lesión.
Múltiples funciones enzimáticas de la quimasa
La quimasa es una proteasa de serina y escinde el lado C-terminal de las proteínas después de aminoácidos aromáticos como Phe, Tyr y Trp en general. La quimasa puede romper el enlace Fe8-His9 del péptido no bioactivo angiotensina I y formar su péptido bioactivo angiotensina II en tejidos de mamíferos, incluidos los humanos (Urata et al., 1990; Takai et al., 1996, 1997). Además, la quimasa escinde enzimáticamente los precursores de la metaloproteinasa de matriz (MMP)-9, el factor de crecimiento transformador (TGF)-β y el colágeno I a sus formas activas (Kofford et al., 1997; Takai et al., 2003; Furubayashi et al., 2008). Además, la función enzimática de la quimasa puede producir factor de células madre (SCF) por escisión enzimática de la forma inactiva unida a la membrana del SCF, que induce la formación de mastocitos maduros a partir de mastocitos inmaduros a través de la estimulación del receptor c-kit (Longley et al., 1997). Por lo tanto, la quimasa tiene múltiples funciones enzimáticas, incluida la activación de la angiotensina II, MMP-9, TGF-β, colágeno I y SCF (Figura 2).
La función enzimática de la quimasa en NASH
La angiotensina II puede promover la esteatosis hepática y la inflamación al aumentar las especies reactivas de oxígeno (ROS) tras la estimulación de los receptores de angiotensina II en modelos animales de NASH (Hirose et al., 2007; Nabeshima et al., 2009). La angiotensina II también indujo fibrosis hepática a través de la inducción de actina de músculo liso α (AMS) en células estrelladas hepáticas (CSH) (Yoshiji et al., 2001). Se ha informado que MMP-9 induce la infiltración de neutrófilos y macrófagos a través de la degradación de matrices intercelulares como vitronectina y fibronectina, lo que resulta en un aumento de la inflamación (Medina et al., 2006). En los pacientes con EHNA, se observó un aumento significativo de la expresión génica MMP-9 en el hígado en comparación con los controles normales (Ljumovic et al., 2004). La sobreexpresión hepática de TGF-β en ratones transgénicos produjo fibrosis hepática grave a través del aumento de la expresión del gen procolágeno I (Casini et al., 1993). Se sabe que tanto la formación de TGF-β como la acumulación de colágeno I inducen fibrosis hepática. La activación de SCF induce aumentos en el número de mastocitos, y su función enzimática puede resultar en un aumento de la actividad quimasa en los tejidos fibróticos (Maruichi et al., 2004). Estas funciones enzimáticas de la quimasa pueden estar implicadas en la esteatosis, inflamación y fibrosis, todas las cuales se observan en el hígado de pacientes con NASH y modelos animales (Figura 2).
Participación de la quimasa en Modelos animales de NASH
La dieta deficiente en metionina y colina (MCD) se ha utilizado ampliamente para inducir un modelo típico de NASH. En los hámsteres alimentados con la dieta MCD, se observaron aumentos significativos de bilirrubina total, triglicéridos y ácido hialurónico en el plasma (Tashiro et al., 2010). Además, se observó acumulación de células inflamatorias y aumento del área de depósitos lipídicos y área fibrótica en el hígado. En este modelo de NASH inducido por la dieta de ECM, la actividad de la quimasa hepática y los factores relacionados, como la angiotensina II, MMP-9 y colágeno I, aumentaron significativamente (Tashiro et al., 2010; Masubuchi et al., 2013). Recientemente, se desarrolló un nuevo modelo de NASH en el que se alimentó a ratas espontáneamente hipertensas 5/Dmcr (SHRSP5/Dmcr) propensas a accidentes cerebrovasculares con una dieta alta en grasas y colesterol (HFC) (Kitamori et al., 2012). Este modelo mostró síntomas de síndrome metabólico que se cree que se parecen clínicamente a los de los pacientes con EHNA (Kitamori et al., 2012). En el modelo NASH inducido por la dieta de HFC, se observó hipertensión e hiperlipidemia, y se detectaron esteatosis severa, fibrosis y acumulación de células inflamatorias en el hígado (Miyaoka et al., 2017). Además, se observó un aumento significativo de la actividad de la quimasa junto con MMP-9, TGF-β y colágeno I en el hígado (Miyaoka et al., 2017). Por lo tanto, parece haber una estrecha relación entre la quimasa y la patogénesis de NASH en modelos animales de NASH.
Efecto del Inhibidor de la Quimasa en Modelos Animales de NASH
Efecto del Inhibidor de la Quimasa en Modelos Animales de NASH
Un inhibidor de la quimasa de molécula baja atenuó significativamente la actividad de la quimasa y disminuyó los niveles de angiotensina II, MMP-9 y colágeno I en el hígado en un modelo de hámster NASH alimentado con dieta de ECM, cuando la administración del inhibidor se inició al mismo tiempo que la dieta de ECM (Tashiro et al., 2010; Masubuchi et al., 2013). El inhibidor de la quimasa previno significativamente la esteatosis hepática, la fibrosis y la acumulación de células inflamatorias en este modelo de NASH (Tashiro et al., 2010; Masubuchi et al., 2013). Se cree que el estrés oxidativo juega un papel en la teoría de los «impactos múltiples» del desarrollo de NASH, y el aumento del marcador de estrés oxidativo malondialdehído se atenuó significativamente en el hígado por el inhibidor de la quimasa (Masubuchi et al., 2013). En un modelo de NASH inducido por la dieta de ECM de hámster, el inhibidor de la quimasa mostró un efecto mejorador cuando se administró en NASH establecido (Masubuchi et al., 2013). Los grados de esteatosis y fibrosis en el hígado se redujeron en comparación con antes de la administración del inhibidor de la quimasa (Masubuchi et al., 2013).
En el hígado de un modelo de NASH inducido por la dieta HFC de rata hipertensa, un inhibidor de la quimasa de molécula baja atenuó los niveles de quimasa, así como MMP-9, TGF-β y colágeno I, que son todos factores asociados a la quimasa (Miyaoka et al., 2017). El inhibidor de la quimasa atenuó significativamente la esteatosis hepática y la fibrosis, y redujo la mieloperoxidasa como marcador de inflamación, particularmente de infiltración de neutrófilos (Miyaoka et al., 2017). En este modelo inducido por la dieta de HFC, la supervivencia del grupo tratado con placebo fue del 0% a las 14 semanas siguientes al inicio de la dieta de HFC, y resultó de insuficiencia hepática grave (Miyaoka et al., 2017). Sin embargo, el grupo tratado con inhibidores de la quimasa, en el que las ratas fueron tratadas con el inhibidor de la quimasa inmediatamente después del inicio de la dieta con HFC, mostró una supervivencia del 100% a las 14 semanas. Además, se notificó una tasa de supervivencia del 50% para las ratas tratadas con el inhibidor de la quimasa a partir de 8 semanas después del inicio de la alimentación con dieta HFC, momento en el que se estableció el NASH (Miyaoka et al., 2017).
Por lo tanto, los inhibidores de la quimasa podrían ser agentes útiles para la prevención y mejora de NASH en modelos animales. Por otro lado, la angiotensina II también promueve indirectamente la inflamación hepática, la esteatosis y la fibrosis a través de aumentos de la expresión génica MMP-9 y TGF-β. Tanto el MMP-9 como el TGF-β están estrechamente implicados en la patogénesis de la EHNA, pero estos factores no son necesariamente inducidos solo por la angiotensina II (Takai et al., 2010). Otros factores, además de la estimulación de la angiotensina II, contribuyen al aumento de la expresión génica MMP-9 y TGF-β (Takai et al., 2010). En tales casos, el antagonista de los receptores de la angiotensina II (ARA II) no es capaz de atenuar las acciones de MMP-9 y TGF-β; sin embargo, un inhibidor de la quimasa podría tener efectos atenuantes a través de la inhibición de la activación de MMP-9 y TGF-β, lo que indica un posible curso de tratamiento para la prevención de la progresión de NASH.
El mecanismo de Inflamación Hepática Atenuado por el inhibidor de la quimasa
El inhibidor de la quimasa fue capaz de reducir la inflamación en modelos de NASH inducidos por dieta de ECM de hámster y HFC de rata (Tashiro et al., 2010; Masubuchi et al., 2013; Miyaoka et al., 2017). El tratamiento con inhibidores de la quimasa atenuó significativamente la actividad de la quimasa en el hígado, así como redujo los niveles de angiotensina II y MMP-9 (Tashiro et al., 2010; Masubuchi et al., 2013; Miyaoka et al., 2017). En HSC, la angiotensina II induce la generación de ROS, como peróxido de hidrógeno y superóxido, a través de la activación de la nicotinamida adenina dinucleótido fosfato (NADPH) oxidasa (De Minicis y Brenner, 2007). El inhibidor de la quimasa produjo reducciones en la expresión génica del componente NADPH oxidasa Rac-1 y del marcador de estrés oxidativo malondialdehído, además de una reducción de los niveles de angiotensina II en un modelo de NASH inducido por ECM de hámster (Masubuchi et al., 2013). El aumento de ROS inducido por angiotensina II promovió la expresión génica de MMP-9 en neutrófilos y macrófagos (Yaghooti et al., 2011; Kurihara et al., 2012). Por lo tanto, el inhibidor de la quimasa inhibe directamente la activación de proMMP-9 a MMP-9 y reduce indirectamente la expresión génica de MMP-9 a través de la disminución de la angiotensina II. MMP-9 escinde los componentes de la matriz extracelular, como la vitronectina y la fibronectina, conduce a la desintegración de la integridad hepática e induce la infiltración de macrófagos y neutrófilos (Medina et al., 2006). En un modelo de NASH inducido por la dieta de HFC, se observó un aumento significativo de la expresión de mieloperoxidasa en macrófagos y neutrófilos en el hígado, y se redujo con el inhibidor de la quimasa (Miyaoka et al., 2017). Por lo tanto, el mecanismo de inflamación atenuado por el inhibidor de la quimasa puede depender de la reducción de los niveles de angiotensina II y MMP-9 en el hígado.
Mecanismo de la esteatosis hepática Atenuado por el Inhibidor de la quimasa
La angiotensina II puede influir en la esteatosis hepática a través de la producción de ROS. En la HSC murina, un inhibidor de la NADPH oxidasa disminuyó significativamente la producción de ROS y un ARA retrasó el desarrollo de esteatosis hepática a través de la atenuación de la producción de ROS (Hirose et al., 2007; Guimarães et al., 2010). En un modelo de ratón NASH inducido por la dieta de ECM, se observó una atenuación significativa de la esteatosis en ratones con deficiencia de receptores de angiotensina II (Nabeshima et al., 2009). Los experimentos in vivo e in vitro mostraron que la angiotensina II reguló al alza la expresión génica de la proteína de unión a elementos reguladores de esteroles (SREBP)-1c y la sintasa de ácidos grasos (FAS), factores importantes en la regulación de la lipogénesis, tras el aumento de ROS (Kim et al., 2001; Hongo et al., 2009). En contraste, ARA atenuó la esteatosis hepática junto con la regulación a la baja de la expresión génica de SREBP-1c y FAS a través de la atenuación de ROS en un modelo de NASH de ratón (Kato et al., 2012). En un modelo de NASH inducido por la dieta de ECM de hámster, se observó una atenuación significativa de la expresión génica de SREBP-1c y FAS después del tratamiento con un inhibidor de la quimasa de molécula baja (Masubuchi et al., 2013). Por lo tanto, el mecanismo de mejora de la esteatosis hepática por el inhibidor de la quimasa puede depender de la reducción de la producción de ROS a través de la reducción de la generación de angiotensina II en el hígado.
El mecanismo de Fibrosis Hepática Atenuado por el Inhibidor de la quimasa
La quimasa puede estar estrechamente asociada con la progresión de la fibrosis tisular, ya que contribuye a la formación de TGF-β a partir del precursor no bioactivo TGF-β, y se sabe que TGF-β induce fuertemente el crecimiento de fibroblastos (Takai et al., 2003; Oyamada et al., 2011). Se sabe que el TGF-β desempeña un papel central en la progresión de la fibrosis en pacientes con EHN a través de la HSC activada (Williams et al., 2000). La inhibición de la función TGF-β a través de la expresión génica y la señalización resultó en una mejora de la fibrosis hepática en modelos experimentales (George et al., 1999; Arias et al., 2003). En un modelo de NASH inducido por la dieta de HFC en ratas, la atenuación de la actividad de la quimasa por el inhibidor de la quimasa dio lugar a reducciones en el nivel de TGF-β y el área fibrótica en el hígado (Miyaoka et al., 2017). Por lo tanto, la reducción del TGF-β por el inhibidor de la quimasa puede contribuir a la prevención de la fibrosis hepática.
La angiotensina II también puede estar implicada en la inducción de fibrosis hepática. La angiotensina II induce la contracción y proliferación de HSC, y también induce la expresión génica de TGF-β en fibroblastos in vitro (Kagami et al., 1994; Bataller et al., 2000). Tanto los niveles de TGF-β como el grado de acumulación de colágeno y lesiones fibróticas se observaron mediante ligadura de vías biliares en ratones de tipo salvaje, sin embargo, estos se atenuaron en ratones con deficiencia de receptores de angiotensina II (Yang et al., 2005). En un modelo de NASH de rata, ARA también atenuó la fibrosis hepática a través de la reducción de la expresión génica de TGF-β (Hirose et al., 2007). También puede existir una relación entre la angiotensina II y la fibrosis hepática distinta de la expresión génica de TGF-β inducida por la angiotensina II. En pacientes con hepatitis C crónica, la ARA redujo la expresión génica de colágeno a través de la expresión génica Rac-1 (Colmenero et al., 2009). Las HSC son reconocidas como las principales células productoras de colágeno en el hígado, y el aumento en la expresión de actina de músculo liso α (SMA) en las HSC induce fuertemente la deposición de matriz extracelular, incluido el colágeno I (De Minicis y Brenner, 2007). La angiotensina II puede inducir la expresión del gen α-SMA en la HSC de ratas. Por el contrario, el bloqueo de la angiotensina II resulta en la atenuación de la fibrosis hepática junto con la reducción de α-SMA (Yoshiji et al., 2001). Aunque no se evaluaron en pacientes con EHNA, las actividades formadoras de quimasa y angiotensina II aumentaron significativamente en las regiones fibróticas del hígado de pacientes con cirrosis, y se observaron correlaciones significativas entre la actividad formadora de quimasa, angiotensina II y fibrosis hepática (Komeda et al., 2008). En un modelo de cirrosis hepática inducida por tetracloruro de hámster, se observaron aumentos significativos en la actividad formadora de quimasa y angiotensina II, que se atenuaron significativamente junto con la cirrosis hepática después del tratamiento con un inhibidor de quimasa de molécula baja (Komeda et al., 2010).
El estabilizador de mastocitos tranilast podría inhibir la activación de los mastocitos, bloqueando la liberación de quimasa y, por lo tanto, previniendo el desarrollo de fibrosis hepática en un modelo NASH inducido por dieta y diabetes de rata (Uno et al., 2008). La quimasa promueve la proliferación de mastocitos a través de la activación del SCF por su función enzimática (Longley et al., 1997). En modelos animales de NASH, el inhibidor de la quimasa redujo el aumento del número de mastocitos en el hígado, lo que resultó en una reducción de la actividad de la quimasa después de la inhibición directa por el inhibidor de la quimasa y una reducción indirecta de la expresión de la quimasa en los mastocitos (Masubuchi et al., 2013; Miyaoka et al., 2017).
Por lo tanto, el inhibidor de la quimasa puede contribuir a la prevención de la fibrosis hepática a través de la inhibición de la activación de TGF-β por inhibición de la quimasa y/o atenuación del nivel de TGF-β a través de la reducción de la angiotensina II y la proliferación de mastocitos.
Conclusión
El síndrome metabólico que comprende obesidad, resistencia a la insulina, hiperlipidemia e hipertensión está estrechamente relacionado con el desarrollo de NASH, y se han realizado ensayos con antidiabéticos, antihiperlipidémicos y antihipertensivos para el tratamiento de NASH. El concepto detrás de estos agentes es atenuar los síntomas del síndrome metabólico (Georgescu et al., 2009; Park et al., 2010; Mahady et al., 2011). Informes anteriores han demostrado que el inhibidor de la quimasa atenúa la inflamación y la fibrosis sin influir en los niveles de glucosa y lípidos en sangre y la presión arterial en modelos animales de diabetes, hiperlipidemia e hipertensión, respectivamente (Inoue et al., 2009; Takai et al., 2014; Zhang et al., 2016). Por lo tanto, el concepto detrás de la inhibición de la quimasa es atenuar la inflamación hepática y la fibrosis de NASH directamente. Proponemos que el síndrome metabólico dirigido a inhibidores de la quimasa es una estrategia potencialmente poderosa para la atenuación de la progresión de NASH.
Contribuciones de autor
ST y DJ: escribieron el manuscrito. Ambos autores leyeron y aprobaron el manuscrito final.
Declaración de Conflicto de Intereses
Los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un conflicto de intereses potencial.