- Introducción
- Presentación del caso
- Caracterización Clínica del Proband y Familiares
- Análisis de Laboratorio y Pruebas de Diagnóstico
- Resultados
- Conclusiones
- Disponibilidad de datos
- Declaración de Ética
- Contribuciones de los autores
- Financiación
- Declaración de Conflicto de Intereses
- Material suplementario
Introducción
Las Distrofias Musculares de las Cinturas de las Extremidades (DMGG) incluyen un grupo heterogéneo de trastornos caracterizados por el desgaste progresivo y la debilidad de los músculos proximales de las cinturas de las extremidades (1, 2). Las DMGG muestran variabilidad intrafamiliar e intrafamiliar, que van desde formas muy leves hasta fenotipos graves, de aparición temprana y de rápida progresión (2). Las LGMD se pueden clasificar como autosómicas dominantes (LGMD1) o recesivas (LGMD2). El primer grupo generalmente tiene una edad adulta de inicio y no son muy frecuentes (<10% de todas las DMGG), mientras que los últimos son más frecuentes (1:15000) (1). Entre las formas recesivas, la LGMD2A (o calpainopatía) es la LGMD más común en todo el mundo, afectando aproximadamente al 30% de todos los casos de LGMD (3). Las señales clínicas de la enfermedad incluyen caminar de puntillas, andar de patitas, dificultad para correr y subir escaleras, alas escapulares. Se observan con frecuencia contracturas articulares, acortamiento del tendón de Aquiles y escoliosis, mientras que los músculos faciales y del cuello no se ven afectados (4). Una hipercemia asintomática (5-80 veces los niveles normales de CPK) en pacientes jóvenes se considera un estadio preclínico de la enfermedad y puede persistir durante varios años (1, 5). La edad de inicio de la debilidad muscular generalmente ocurre a los 15 años, aunque puede aparecer a edades anteriores (<12 años) o posteriores (>30 años). La progresión de la enfermedad puede conducir a la pérdida de la deambulación, insuficiencia respiratoria y reducción de la capacidad vital pulmonar en las etapas avanzadas (6). La afectación cardíaca (trastornos del ritmo cardíaco, trastornos de la conducción cardíaca, disfunción de la eyección del ventrículo izquierdo) solo se notifica ocasionalmente (6, 7).
El diagnóstico de calpainopatía se confirma mediante la detección de mutaciones patógenas en CAPN3 (15q15.1) (4), codificando diferentes transcripciones unidas alternativamente. Sin embargo, la transcripción completa se expresa principalmente en tejido muscular (8). La proteína codificada (CAPN3) es un miembro de la familia de cisteína proteasas no lisosómicas dependientes de Ca++. En el músculo, CAPN3 participa en la» remodelación de sarcómeros», que es esencial para la adaptación y el crecimiento muscular en respuesta a las demandas funcionales y metabólicas. Hasta la fecha, se han descrito más de 490 mutaciones patógenas a lo largo de CAPN3, la mayoría de las cuales son cambios de un solo nucleótido (4). Las mutaciones en CAPN3 se han asociado con anomalías mitocondriales, insuficiencia de crecimiento, aumento del estrés oxidativo y desorganización de los sarcómeros, que en conjunto contribuyen a que el músculo no pueda soportar cargas, causando degeneración de miofibras y desgaste muscular (8). El diagnóstico de calpainopatía puede ser difícil debido a la heterogeneidad genética y la no especificidad del patrón clínico e instrumental. De hecho, la distribución de la debilidad/atrofia muscular, la hipertrofia/pseudo-hipertrofia y las contracturas tendinosas se comparten muy a menudo con otras DMGG o trastornos neuromusculares. El patrón de biopsia muscular en la calpainopatía también es generalmente inespecífico, desde anomalías musculares leves hasta cambios distróficos graves. Además, los marcadores inmunohistoquímicos/bioquímicos generalmente no son confiables: la señal de calpaína puede ser normal incluso en presencia de una proteína no funcional, y viceversa, puede reducirse incluso en otras distrofias musculares diferentes de la calpainopatía.
Por lo tanto, se recomienda realizar un diagnóstico diferencial para proporcionar resultados precisos y confiables. Con este fin, se han desarrollado métodos de pruebas genéticas moleculares para confirmar el diagnóstico de la calpainopatía, que incluyen un panel multigénico, un análisis genómico extenso (secuenciación del exoma/genoma) y un análisis de un solo gen (secuenciación directa) (9). La implementación de la Secuenciación de Próxima Generación (NGS) fue útil para generar datos informativos que se aplicarían con fines diagnósticos, predictivos o terapéuticos (10-12). Los paneles de genes NGS se basan en el análisis de un conjunto de genes asociados con una enfermedad específica o un grupo de trastornos relacionados, que se caracterizan por la heterogeneidad genética y fenotípica. Los paneles NGS también pueden ser útiles para detectar fenotipos mezclados o complejos, que resultan de la herencia de más de un defecto genético de los padres (13). En general, los pacientes que presentan fenotipos complejos, que son negativos a las mutaciones conocidas en paneles genéticos de diagnóstico diseñados a medida y que requieren un diagnóstico diferencial amplio, son elegibles para la secuenciación completa del exoma o genoma. La secuenciación completa del exoma/genoma es un enfoque más costoso en términos de gestión de datos, interpretación y costos analíticos, pero aumenta la probabilidad de proporcionar un diagnóstico molecular a un presunto trastorno genético (13). En cuanto a las LGMD, se recomiendan encarecidamente paneles NGS dedicados para garantizar altas tasas de diagnóstico, cobertura óptima, sensibilidad y especificidad de las pruebas clínicas (9). Por lo tanto, los paneles NGS representan uno de los mejores sistemas para facilitar el diagnóstico diferencial, identificar nuevas mutaciones causales y aclarar las correlaciones genotipo-fenotipo (14, 15).
En este manuscrito, se reporta el caso de un paciente afectado por LGMD2A, para el cual la aplicación del panel NGS permitió no solo confirmar el diagnóstico de calpainopatía (mutaciones en CAPN3), sino también identificar una mutación adicional y novedosa en el gen LMNA asociada a miocardiopatía dilatada. Dados estos resultados, el análisis se extendió a los familiares del proband para proporcionar una interpretación más completa de los datos analíticos en relación con los fenotipos patológicos.
Presentación del caso
Caracterización Clínica del Proband y Familiares
El inicio de la enfermedad en el proband fue referido a los 10 años de edad, cuando relató la presencia de hipertrofia de pantorrillas con caminar de puntillas, dificultades para correr, subir escaleras y ponerse de pie. Los síntomas mostraron un empeoramiento lento pero progresivo, con compromiso posterior del miembro superior proximal. A la edad de 13 años, se descubrieron niveles altos de CPK (~8,000 UI/L), nunca asociados con mioglobinuria. Sucesivamente, el paciente se sometió a varias investigaciones neuromusculares en varios centros especializados. Se realizaron dos biopsias musculares, mostrando un cuadro distrófico clásico (fibras hipertróficas/atróficas, núcleos internos, fibras necróticas, aumento del tejido conectivo) con características inespecíficas. Como era de esperar, los estudios de inmunoquímica e inmunoblots no fueron concluyentes indicando señales anormales / reducidas de α, β, γ sarcoglicano, β-distroglicano y distrofina solo en fibras necróticas (la inmunoquímica repetida resultó normal, también para laminina); los inmunoblots para distrofina y calpaína revelaron señales normales. Sin embargo, se sabe que el inmunoblot calpain-3 no es completamente sensible para el diagnóstico de LGMD2A, ya que el 20-30% de los casos muestran una cantidad normal de proteína (1, 16-18). Debido a la presentación clínica (hiperCKmia, hipertrofia/pseudo-hipertrofia de pantorrillas), se descartaron en primer lugar distrofinopatías. Además, el análisis de los genes DMD (Xp21), FKRP (19q13.32) y DYSF (2p13.2) no reveló mutaciones patógenas. La paciente llegó a nuestra observación a la edad de 30 años, presentando compromiso axial y de cintura tanto en el miembro superior como en el inferior, con una marcha significativa al andar y escápulas aladas. En las extremidades inferiores, la debilidad era prominente en los músculos cuádriceps y glutei, que eran significativamente ipo-atróficos, junto con la evidencia de músculos de pantorrillas «aparentemente hipertróficos» (pseudo-hipertrofia) (Figura 1). No se observaron calambres, mialgias y ondulaciones. Los exámenes neumológicos completos (espirometría y polisomnografía) y cardiológicos (ecografía, ECG Holter de 24 horas, resonancia magnética cardíaca, inspección física cardiológica completa con antecedentes médicos específicos y análisis de reflejos cardiovasculares) no revelaron ninguna anomalía significativa. También descartamos la deficiencia de maltasa ácida (análisis de actividad enzimática normal en linfocitos/leucocitos). La resonancia magnética muscular (RM) mostró una afectación prominente de la cintura escapular, los músculos paravertebrales, los músculos del compartimento posterior del muslo (con relativa preservación de los músculos sartorius y gracilis) y de la pierna (en particular, el gastrocnemius medialis y el sóleo) (Figura 2). Este patrón de compromiso ya se ha descrito en la literatura sobre la imagen de resonancia magnética en la calpainopatía (19, 20). Además, el estudio de resonancia magnética mostró que los músculos de la pantorrilla eran efectivamente atróficos y se caracterizaban por infiltración de grasa, causando un «agrandamiento muscular».»La presentación clínica, la edad de inicio, la tasa de progresión, la distribución de la debilidad muscular y los hallazgos de la RMN en el proband fueron totalmente consistentes con un diagnóstico de LGMD2A ampliamente descrito en la literatura (4, 21, 22).FIGURA
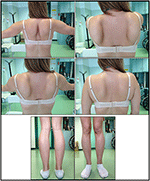
Gráfico 1 Características clínicas del probanda: escápula alada y combinación de atrofia de muslos y pseudo-hipertrofia de pantorrillas.
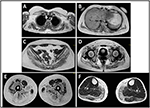
Figura 2. Resonancia magnética de los músculos probandos: afectación de la faja de las extremidades superiores y de los músculos paravertebrales (A,B), afectación de la faja de las extremidades inferiores y del compartimento posterior del muslo (C–E, principalmente los glúteos) con preservación relativa de sartorius y gracilis, y afectación del compartimento posterior de la pierna (F, principalmente gastrocnemius medialis/sóleo).
La evaluación de los familiares del proband no reveló antecedentes de enfermedad neuromuscular ni evidencia de consanguinidad entre los padres. Sin embargo, la madre del probanda, a la edad de 55 años, presentó 2 episodios sincopales por los que se le diagnosticó bradicardia idiopática (hasta 40 lpm por noche). Extensas investigaciones cardiológicas en la madre del probanda diagnosticaron un bloqueo auriculoventricular (AV) sintomático después de la aparición de varios episodios sincopales/lipotimia. El análisis mostró la presencia de un bloqueo AV básico de primer grado, con varios períodos de bradicardia severa, en su mayoría nocturna y a menudo sintomática, asociada a algunas fases de bloqueo AV de segundo grado, tanto de tipo 1 como 2, y algunas fases de disociación AV completa nocturna. La ecocardiografía y la prueba ergométrica no revelaron alteraciones significativas, excepto en el foramen oval permeable. La madre no presentó ningún otro síntoma clínico, condición predisponente o factor de riesgo. Dado su cuadro clínico, a la madre del proband se le implantó PMK. In addition, family history revealed that the grandmother and the bisabuelo of the proband were also implanted with PMK at the age of 55 and 30 years, respectively. Moreover, the brother of the grandmother of the proband died at 51 for severe dilated cardiomyopathy. También se realizaron exámenes cardiológicos al padre y al tío materno del proband, que resultaron completamente inafectados.
El presente estudio fue aprobado por el comité de ética de la Fundación Santa Lucía y se realizó de acuerdo con la Declaración de Helsinki. Todos los participantes dieron su consentimiento informado firmado para el análisis genético y, en este sentido, también dieron su consentimiento para la publicación de este reporte de caso.
Análisis de Laboratorio y Pruebas de Diagnóstico
Se extrajo ADN genómico de sangre periférica (400 µL) utilizando el Kit de Extracción de ADN Sanguíneo MagPurix y el Sistema de Extracción Automática MagPurix (Resnova) de acuerdo con las instrucciones del fabricante. Las muestras se secuenciaron utilizando el Sistema Ion PGM y el Panel Personalizado Ion Ampliseq de Alta Especificidad (Thermo Fisher Scientific). El tamaño del panel era de 129.13 Kb, que se espera que tamice ~99,72% del panel total con una cobertura mínima de 20X. El panel incluyó 18 genes, que se seleccionaron utilizando literatura científica, GeneReviews (www.ncbi.nlm.nih.gov/books/NBK1116/) y frecuencia de variantes patógenas en la población general. En el cuadro 1 se ha resumido una descripción detallada del grupo de SGN.
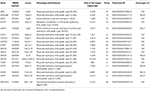
Cuadro 1 Panel NGS personalizado utilizado para el diagnóstico de LGMD.
La construcción de bibliotecas se realizó con los kits de bibliotecas Ion AmpliSeq™ 2.0. Se utilizaron aproximadamente 10 ng/µl de ADN inicial para reacciones de PCR múltiple. Sucesivamente, se realizaron dos pasos de purificación (utilizando AMPure XP, Beckman Coulter) para eliminar contaminantes no deseados y se realizó una PCR final. Los pasos de amplificación y enriquecimiento de la plantilla se realizaron con el kit Ion PGM Hi-Q OT2-400, el sistema Ion OneTouch 2 y el sistema Ion OneTouch ES (Thermo Fisher Scientific). Las muestras se procesaron con el Kit de secuenciación Ion PGM Hi-Q (400 bp, Thermo Fisher Scientific) y se ejecutaron con el chip Ion 316 v2 (se requieren 850 flujos) y el secuenciador Ion PGM (Thermo Fisher Scientific). Los resultados se analizaron utilizando el Ion Reporter 4.6 (Thermo Fisher Scientific) y el Visor Integrado del Genoma (IGV). La interpretación de las variantes genéticas fue realizada por Human Gene Mutation Database (HGMD), Leiden Open Variation Database (LOVD), ClinVar y ExAC. El efecto funcional de las variantes detectadas se evaluó mediante herramientas predictivas bioinformáticas, como el Probador de mutaciones, Varsome, SIFT, PolyPhen 2, SMART, Human Splicing Finder (HSF). Secuenciación directa (BigDye Terminator v3.1, BigDyeX Terminator y ABI3130, Applied Biosystems) se realizó para confirmar variantes genéticas y secuenciar regiones codificantes genómicas con una cobertura <20X (LMNA, Chr1:156106052-156106076; DYSF, Chr2:71753352-71753502, Chr2:71776385-71776640; SGCB, Chr4:52904225-52904560; SGCA, Chr17:48243242-48243570).
Resultados
El análisis NGS del proband reveló 3 variantes heterocigotas (Figura Suplementaria 1). Dos variantes fueron localizados en CAPN3, es decir, NM_000070.2 (CAPN3): c.550delA (p.Thr184Argfs) y c).1813G>C (p.Val605Leu) en los exones 4 y 16, respectivamente. La primera es la deleción de un solo nucleótido y es la variante patógena más común (75% de los casos) en los países europeos (1). Como era de esperar, el análisis bioinformático clasificó el c. 550delA (rs80338800) como una variante de pérdida de función (p. Thr184Argfs), causando un cambio de fotogramas del marco de lectura abierto. Las herramientas predictivas (Probador de mutaciones, HSF, Varsome, PoliFeno 2, TAMIZ) describieron la variante como perjudicial. Además, SMART reveló que el producto de proteína alterado carece del dominio calpain 3 y los tres motivos de «manos EF». El primer dominio está involucrado en las vías de señalización de la Calpaína, mientras que las manos EF son esenciales para la activación dependiente de Ca++de la proteína (4).
En relación con c. 1813G> C, es una nueva variante sin sentido (p. Val605Leu) que se ha predicho que es perjudicial (por Probador de Mutaciones, HSF, Varsome, PoliFeno 2, TAMIZ) debido a una alteración potencial del empalme. Esta variante no está anotada en la literatura ni en las bases de datos en línea y no se ha encontrado en 200 sujetos de control. Desafortunadamente, el análisis de SMART tool no arrojó resultados significativos, ya que la variante se encuentra dentro de un dominio no caracterizado. El análisis de la segregación de CAPN3 mostró que la madre y el tío materno de ambos eran heterocigotos para c.550delA, mientras que el padre era portador de c.1813G>C.
además, NGS análisis en el proband reveló la presencia de una nueva variante en LMNA, es decir, NM_170707 (LMNA): c.550C>T (p.Gln184*). Se ha pronosticado que la variante mencionada anteriormente es una variante nula, aún no se ha descrito en la literatura ni en las bases de datos en línea y no se ha encontrado en 200 sujetos de control. Las herramientas predictivas (Probador de mutaciones, HSF, Varsome, PoliFeno 2, TAMIZ) describieron un efecto patógeno de esta variante en el producto proteico. SMART tool informó que la proteína LMNA alterada carece del dominio de filamentos, lo que es esencial para mantener su estructura y función. El análisis de segregación destacó la presencia de la c.Variante 550C>T solo en la madre, lo que sugiere una posible asociación con su sintomatología cardíaca y su historia familiar de enfermedad.
De acuerdo con los criterios establecidos por los Estándares y Directrices del American College of Medical Genetics (ACMG) (23), c. 1813G>C (CAPN3) se puede describir como una variante patógena probable teniendo en cuenta que se encuentra en un dominio crítico sin variación benigna (PM1); está ausente en las bases de datos ExAc, GnomAD y 1000 Genome Browser (PM2); considerando el modelo recesivo de herencia, se ha detectado en trans con una variante patógena (PM3); múltiples líneas de evidencia computacional apoyan un efecto perjudicial sobre el gen o el producto del gen (PP3). En cuanto a la clasificación clínica de c. 550C>T (LMNA) por ACMG, se puede designar como una variante patógena, ya que es una variante nula que causa pérdida de función (p. Gln184*) en LMNA (PVS1); está ausente en las bases de datos ExAc, GnomAD y 1000 Genome Browser (PM2); se encuentra en un dominio crítico sin variación benigna (PM1); múltiples líneas de evidencia computacional apoyan un efecto perjudicial sobre el gen o el producto del gen (PP3).
Conclusiones
Este reporte de caso presentó un paciente afectado por DMGG, que se encontró portador de mutaciones no solo en CAPN3 (c. 550delA y c. 1813G>C), sino también en LMNA (c. 550C> T). Estos resultados explican el fenotipo neuromuscular del probanda debido a mutaciones en CAPN3 y destacan un riesgo potencial de trastornos cardiovasculares debido a la presencia de la variante en el LMNA y su historia familiar positiva. En vista de estos resultados, los miembros de la familia fueron sometidos al análisis de segregación. En particular, la madre resultó ser portadora de CAPN3_c.550delA y LMNA_c.550C>T, respectivamente. La madre puede ser considerada portadora sana de la mutación patogénica CAPN3_c.550delA asociada a la calpainopatía. Sin embargo, la presencia de una variante novedosa en el LMNA puede explicar su patología cardiovascular (bradicardia y episodios sincopales). La ausencia de sintomatología neuromuscular en la madre y el cuadro clínico peculiar del proband excluyeron una posible asociación de LMNA_c.550C> T con fenotipo neuromuscular, especialmente en relación con la distrofia Muscular de Emery-Dreifuss (EMD). De hecho, la EMD afecta específicamente a los músculos vastos lateralis y bíceps braquiales que están relativamente preservados en LGM2A (24, 25).
El tío materno era heterocigotos sólo para el CAPN3_c.550delA, y, como era de esperar, no muestran ningún neuromuscular o problemas cardiovasculares. El padre llevó la novela CAPN3_c.1813G>C y, por lo tanto, no se vio afectado. En general, el análisis de segregación confirmó la herencia de las tres mutaciones del probanda de sus familiares y destacó una familiaridad para la miocardiopatía que no se puede descuidar (Figura 3).FIGURA
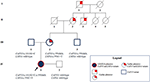
Gráfico 3 Pedigrí que muestra la familiaridad positiva para el fenotipo cardíaco heredado por linaje materno y la transmisión de las variantes CAPN3 y LMNA a través de los miembros de la familia.
En conjunto, estos datos plantean algunas consideraciones importantes. En primer lugar, el panel de NGS en este paciente fue crítico para llegar al diagnóstico molecular de calpainopatía, detectando una mutación conocida y una segunda variante nueva pronosticada como patógena. En segundo lugar, el panel NGS permitió la identificación de la nueva variante LMNA_c.550C>T en el probando. Este resultado fue consistente con los antecedentes familiares positivos y con los datos de segregación, explicando la enfermedad cardiovascular en la madre y, lo que es más importante, recomendando un seguimiento cardiológico más específico en el probanda. Es importante señalar que las manifestaciones cardiológicas ocurrieron en la madre más tarde (55 años), por lo que podemos suponer que el proband podría estar todavía en una «fase cardiológica asintomática».»Sin embargo, no se puede excluir una expresión fenotípica variable de mutación del LMNA en el probanda en comparación con la madre. Todos estos datos subrayan la importancia de un enfoque integrado entre médicos y genetistas, para la interpretación correcta de los resultados, el asesoramiento genético adecuado y, eventualmente, el manejo clínico y el seguimiento. En cuanto al asesoramiento genético y familiar, también se realizó el análisis NGS en la pareja del proband, con el fin de estimar el riesgo reproductivo para la pareja. El compañero fue negativo en todos los 18 genes analizados, lo que significa que tiene un riesgo residual de 1/650 de ser portador sano de mutaciones causantes de la DMGG, considerando que la prueba es sensible al 84%. Teniendo en cuenta el perfil genético de la prueba y la sensibilidad de la prueba NGS, el riesgo residual para la pareja de tener un hijo afectado con calpainopatía es de 1/1300. Por otro lado, las variantes patógenas del LMNA se transmiten de acuerdo con un patrón autosómico dominante, lo que significa que el probanda tiene un 50% de probabilidad de tener hijos heterocigotos. Sin embargo, el cuadro clínico de la descendencia no se puede predecir con certeza, ya que el LMNA_c.550C>T es una variante novedosa y su impacto funcional en el fenotipo es aún desconocido.
En conclusión, este informe de caso destaca la utilidad clínica de los paneles NGS para proporcionar un diagnóstico preciso de LGMD2A y describir fenotipos complejos y comorbilidades que se originan en la herencia de diferentes mutaciones en múltiples genes. Sin embargo, la aplicación de SGN en la práctica clínica siempre debe combinarse con un asesoramiento pre y postgenético para proporcionar una explicación clara de los resultados, las posibles implicaciones en el fenotipo de los pacientes, el riesgo de recurrencia dentro de la familia, así como para explicar posibles hallazgos inesperados.
Disponibilidad de datos
No se generaron ni analizaron conjuntos de datos para este estudio.
Declaración de Ética
Este estudio se realizó de acuerdo con las recomendaciones del comité de ética de la Fundación Santa Lucía con el consentimiento informado por escrito de todos los sujetos. Todos los sujetos dieron su consentimiento informado por escrito de acuerdo con la Declaración de Helsinki. El protocolo fue aprobado por el comité de ética de la Fundación Santa Lucía.
Contribuciones de los autores
RC, CSt, VC, GC, RG, GPa, SC, CP y JM hicieron contribuciones para la adquisición de datos, el análisis y la interpretación de datos. CSt, RC, GC, RG, GM y EG han participado en la redacción del manuscrito. SZ, GPr, CSa y SS han participado en la adquisición de datos clínicos. RC, CSt, CV, GC, RG, GPa, SC, CP, JM, SZ, GPr, GM, CSa, SS y EG han dado la aprobación final de la versión que se publicará.
Financiación
Este trabajo cuenta con el apoyo del Ministerio Nacional de Salud 5X 2016 2.
Declaración de Conflicto de Intereses
Los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un conflicto de intereses potencial.
Material suplementario
El Material Suplementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. Diagnóstico genético y de proteínas de la distrofia muscular de la faja de las extremidades tipo 2A: el rendimiento y las trampas. Nervio Muscular. (2015) 52:163–73. doi: 10.1002 / mus.24682
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
2. Nigro V, Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update (en inglés). Acta Myol. (2014) 33:1–12.
Resumen de PubMed | Google Scholar
3. Richard I, Hogrel JY, Stockholm D, Payan CAM, Fougerousse F, Eymard B, et al. Historia natural de LGMD2A para delinear medidas de resultados en ensayos clínicos. Ann Clin Transl Neurol. (2016) 3:248–65. doi: 10.1002 / acn3.287
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
4. Angelini C, Fanin M. Calpainopatía. En: Adam MP, Ardinger HH, Pagon RA editors. GeneReviews®. Seattle, WA: Universidad de Washington (2017) 1-30.
Resumen de PubMed | Google Scholar
5. Kyriakides T, Angelini C, Schaefer J, Sacconi S, Siciliano G, Vilchez JJ, et al. Directrices de la EFNS sobre el abordaje diagnóstico de la hipercemia pauci o asintomática. Eur J Neurol. (2010) 17:767–73. doi: 10.1111 / j. 1468-1331. 2010. 03012.x
Texto completo de CrossRef / Google Scholar
6. Mori-Yoshimura M, Segawa K, Minami N, Oya Y, Komaki H, Nonaka I, et al. Disfunción cardiopulmonar en pacientes con distrofia muscular de las cinturas de las extremidades 2A. Nervio muscular. (2017) 55:465–9. doi: 10.1002 / mus.25369
Texto completo de CrossRef / Google Scholar
7. Miocardiopatía de Okere A, Reddy SS, Gupta S, Shinnar M. A en un paciente con distrofia muscular de la faja de las extremidades tipo 2A. (2013) 6:e12–13. doi: 10.1161 / CIRCHEARTFAILURE.112.971424
Texto completo de CrossRef / Google Scholar
8. Kramerova I, Ermolova N, Eskin A, Hevener A, Quehenberger O, Armando AM, et al. La falta de regulación ascendente de la transcripción de genes necesarios para la adaptación muscular subyace a la distrofia muscular de la faja de las extremidades 2A (calpainopatía). Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093/hmg / ddw086
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
9. Thompson R, Straub V. Distrofias musculares de la faja de las extremidades-colaboraciones internacionales para la investigación traslacional. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038/nrneurol.2016.35
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
10. Strafella C, Caputo V, Galota MR, Zampatti S, Marella G, Mauriello S, et al. Aplicación de la medicina de precisión en enfermedades neurodegenerativas. Neurol Delantero. (2018) 9:701. doi: 10.3389 / fneur.2018.00701
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
11. Cascella R, Strafella C, Caputo V, Errichiello V, Zampatti S, Milano F, et al. Hacia la aplicación de la medicina de precisión en la Degeneración Macular Relacionada con la Edad. Prog Retin Eye Res. (2017) 63: 132-46. doi: 10.1016 / j. preteyeres.2017.11.004
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
12. Cascella R, Strafella C, Longo G, Manzo L, Ragazzo M, De Felici C, et al. Evaluar el riesgo individual de DMAE con asesoramiento genético, antecedentes familiares y pruebas genéticas. Ojo. (2017) 32:446–50. doi: 10.1038/ojo.2017.192
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
13. Adams DR, Eng CM. Secuenciación de última generación para diagnosticar posibles trastornos genéticos. N Engl J Med. (2019) 379:1353–62. doi: 10.1056 / NEJMc1814955
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
14. Angelini C. Enfermedad neuromuscular. Diagnóstico y descubrimiento de la distrofia muscular de las cinturas de las extremidades. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038/nrneurol.2015.230
Texto completo de CrossRef / Google Scholar
15. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, et al. Uso de la secuenciación del exoma completo para el diagnóstico de la distrofia muscular de las cinturas de las extremidades: resultados y lecciones aprendidas. JAMA Neurolog. (2015) 72:1424–32. doi: 10.1001 / jamaneurol.2015.2274
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
16. Milic A, Daniele N, Lochmüller H, Mora M, Comi GP, Moggio M, et al. Un tercio de las biopsias de LGMD2A tienen actividad proteolítica normal de calpaína 3 determinada por un ensayo in vitro. Desorden Neuromuscular. (2007) 17:148–56. doi: 10.1016 / j. nmd.2006.11.001
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
17. Lanzillo R, Aurino S, Fanin M, Aguennoz M, Vitale F, Fiorillo C, et al. Calpainopatía de inicio temprano con nivel 3 de calpaína normal no funcional. Dev Med Child Neurol. (2006) 48:304–6. doi: 10.1017 / S001216220600065X
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
18. Talim B, Ognibene A, Mattioli E, Richard I, Anderson LV, Merlini L. Expresión normal de calpaína en distrofia muscular de la faja de la extremidad confirmada genéticamente tipo 2A.Neurología. (2001) 56:692–3. doi: 10.1212 / wnl.56.5.692-a
Texto completo de CrossRef / Google Scholar
19. Díaz-Manera J, Llauger J, Gallardo E, Illa I. Resonancia magnética muscular en distrofias musculares. Acta Myol. (2015) 34:95–108.
Resumen de PubMed | Google Scholar
20. Mercuri E, Bushby K, Ricci E, Birchall DR, Pane M, Kinali M, et al. Hallazgos de resonancia magnética muscular en pacientes con distrofia muscular de la faja de las extremidades con deficiencia de calpaína 3 (LGMD2A) y contracturas tempranas. Desorden Neuromuscular. (2005) 15:164–71. doi: 10.1016 / j. nmd.2004.10.008
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
21. Magri F, Nigro V, Angelini C, Mongini T, Mora M, Moroni I, et al. El registro italiano de distrofia muscular de la faja de las extremidades: frecuencia relativa, características clínicas y diagnóstico diferencial. Nervio Muscular. (2017) 55:55–68. doi: 10.1002 / mus.25192
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
22. Chae J, Minami N, Jin Y, Nakagawa M, Murayama K, Igarashi F, et al. La calpaína 3 mutaciones en el gen: genético y clínico-patológicas de los hallazgos en la distrofia muscular de cinturas. Desorden Neuromuscular. (2001) 11:547–55. doi: 10.1016 / S0960-8966(01)00197-3
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
23. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038 / gim.2015.30
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
24. Bonne G, Mercuri E, Muchir A, Urtizberea A, Bécane HM, Recan D, et al. Espectro genético clínico y molecular de la distrofia muscular autosómica dominante de Emery-Dreifuss debida a mutaciones del gen lamin A / C. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170::AYUDA-ANA6>3.0.CO; 2-J
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
25. Hong JS, Ki CS, Kim JW, Suh YL, Kim JS, Baek KK, et al. Arritmias cardíacas, miocardiopatía y distrofia muscular en pacientes con distrofia muscular de Emery-Dreifuss y distrofia muscular de las cinturas de las extremidades tipo 1B. J (2005) 20:283–90. doi: 10.3346 / jkm.2005.20.2.283
Texto completo de CrossRef / Google Scholar