- Einleitung
- Multipuls Funktionen von Chymase
- Chymase in Mastzellen
- Multiple enzymatische Funktionen von Chymase
- Enzymatische Funktion der Chymase in NASH
- Beteiligung von Chymase an NASH-Tiermodellen
- Wirkung von Chymasehemmer in NASH-Tiermodellen
- Wirkung von Chymasehemmer in NASH-Tiermodellen
- Mechanismus der durch Chymasehemmer abgeschwächten Leberentzündung
- Mechanismus der durch Chymasehemmer abgeschwächten Lebersteatose
- Mechanismus der durch Chymasehemmer abgeschwächten Leberfibrose
- Schlussfolgerung
- Autorenbeiträge
- Interessenkonflikterklärung
Einleitung
Die nichtalkoholische Fettlebererkrankung (NAFLD) wurde als häufigste Form der Lebererkrankung anerkannt (Angulo, 2002; Clark et al., 2002). Die nichtalkoholische Steatohepatitis (NASH) ahmt trotz fehlender Trinkgeschichte eine alkoholische Hepatitis nach (Ludwig et al., 1980). NAFLD und NASH sind mit metabolischem Syndrom assoziiert, das auf Fettleibigkeit, Insulinresistenz, Hyperlipidämie und Bluthochdruck zurückzuführen ist. NAFLD gilt als die häufigste Lebererkrankung und stellt sich typischerweise als einfache Lebersteatose dar (Tiniakos et al., 2010). Im Gegensatz dazu ist NASH durch schwere Steatose, lobuläre Entzündung und Fibrose der Leber gekennzeichnet (Powell et al., 1990; Bertot und Adams, 2016). Obwohl der Mechanismus, der für die Entwicklung von NASH verantwortlich ist, unklar bleibt, wird vorgeschlagen, dass NASH durch einen Multiple-Hit-Prozess verursacht wird, mit Lebersteatose als erstem Hit und nachfolgenden Treffern wie Entzündungen, oxidativem Stress und Endotoxinen (Tilg und Moschen, 2010). NASH ist eng mit dem metabolischen Syndrom verwandt, und mehrere klinische Studien haben die therapeutische Behandlung von NASH untersucht, indem sie sich auf die Symptome von Diabetes, Hyperlipidämie und Bluthochdruck konzentrierten (Georgescu et al., 2009; Park et al., 2010; Mahady et al., 2011). Es wurden jedoch keine allgemein akzeptierten therapeutischen Mittel etabliert.
Chymase kann an der Pathogenese der Leberfibrose beteiligt sein. Die Chymaseaktivität war in der Leber von Patienten mit Fibrose oder Zirrhose signifikant erhöht und es bestand eine signifikante Korrelation zwischen dem Chymasespiegel und dem Grad der Fibrose (Komeda et al., 2008). Obwohl eine erhöhte Chymaseaktivität bei Patienten mit NASH nicht berichtet wurde, wurde sie in Tiermodellen von NASH beobachtet (Tashiro et al., 2010; Masubuchi et al., 2013; Miyaoka et al., 2017). Im Gegensatz dazu führte die Hemmung der Chymase mit niedermolekularen Inhibitoren zu einer signifikanten Reduktion von Entzündungen, Steatose und Fibrose in NASH-Modellen von Ratten und Hamstern (Tashiro et al., 2010; Masubuchi et al., 2013; Miyaoka et al., 2017). Diese Ergebnisse deuten darauf hin, dass Chymase während der Entwicklung und des Fortschreitens von NASH an Entzündungen, Steatosen und Fibrosen beteiligt sein kann (Abbildung 1).
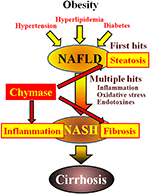
ABBILDUNG 1. NAFLD und NASH sind durch Fettleibigkeit, Insulinresistenz, Hyperlipidämie und Bluthochdruck mit dem metabolischen Syndrom verbunden. Es wird angenommen, dass sich NASH über einen „Multiple-Hit“ -Prozess mit Lebersteatose als „erstem Treffer“ und nachfolgenden Treffern wie Entzündungen, oxidativem Stress und Endotoxinen entwickelt und durch schwere Steatose, Entzündung und Fibrose gekennzeichnet ist. Chymase kann am Fortschreiten von Steatose, Entzündung und Fibrose in der Leber beteiligt sein.
Multipuls Funktionen von Chymase
Chymase in Mastzellen
Chymase (EC 3.4.21.39) wird in den sekretorischen Granula von Mastzellen exprimiert. Chymase wird als inaktive Prochymase innerhalb sekretorischer Granula produziert und benötigt Dipeptidylpeptidase I (DPPI) zur Aktivierung. DPPI ist eine Thiolproteinase und sein optimaler pH-Wert beträgt 6,0. Der optimale pH-Wert stimmt mit der vorgeschlagenen Funktion von DPPI zur Aktivierung der Prochymase überein, da der pH-Wert innerhalb der sekretorischen Granula bei pH 5,5 reguliert wird (De Young et al., 1987) (Abbildung 2). Chymase hat jedoch bei diesem pH-Wert keine enzymatische Aktivität in Mastzellen, da der optimale pH-Wert für Chymase zwischen 7 und 9 liegt (Takai et al., 1996, 1997). Nach der Aktivierung von Mastzellgranulaten durch Reize wie Entzündungen und Verletzungen wird Chymase freigesetzt und zeigt eine enzymatische Funktion bei seinem optimalen pH-Wert von 7,4 (Abbildung 2).
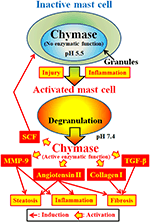
ABBILDUNG 2. Chymase wird in den sekretorischen Granula inaktiver Mastzellen gespeichert. Der pH-Wert innerhalb des Granulats wird bei pH 5,5 gehalten, ein Zustand, in dem Chymase keine enzymatische Aktivität aufweist. Chymase zeigt seine enzymatischen Funktionen, wie Bildung von Angiotensin II, MMP-9, TGF-β, Kollagen I und SCF, nach Freisetzung aus Mastzellgranulat, nach Aktivierung durch Entzündung und Verletzung.
Multiple enzymatische Funktionen von Chymase
Chymase ist eine Serinprotease und spaltet die C-terminale Seite von Proteinen nach aromatischen Aminosäuren wie Phe, Tyr und Trp im Allgemeinen. Chymase kann die Phe8–His9-Bindung des nicht bioaktiven Peptids Angiotensin I spalten und sein bioaktives Peptid Angiotensin II in Säugetiergeweben einschließlich Mensch bilden (Urata et al., 1990; Takai et al., 1996, 1997). Darüber hinaus spaltet Chymase enzymatisch die Vorläufer der Matrix-Metalloproteinase (MMP) -9, des transformierenden Wachstumsfaktors (TGF) -β und des Kollagens I in ihre aktiven Formen (Kofford et al., 1997; Takai et al., 2003; Furubayashi et al., 2008). Darüber hinaus kann die enzymatische Funktion der Chymase durch enzymatische Spaltung der inaktiven membrangebundenen Form von SCF Stammzellfaktor (SCF) erzeugen, der über die Stimulation des c-kit-Rezeptors die Bildung reifer Mastzellen aus unreifen Mastzellen induziert (Longley et al., 1997). Somit hat Chymase mehrere enzymatische Funktionen, einschließlich der Aktivierung von Angiotensin II, MMP-9, TGF-β, Kollagen I und SCF (Abbildung 2).
Enzymatische Funktion der Chymase in NASH
Angiotensin II kann hepatische Steatose und Entzündung fördern, indem es reaktive Sauerstoffspezies (ROS) nach Stimulation von Angiotensin-II-Rezeptoren in tierischen NASH-Modellen erhöht (Hirose et al., 2007; Nabeshima et al., 2009). Angiotensin II induzierte auch Leberfibrose durch Induktion von α-glattem Muskelaktin (SMA) in hepatischen Sternzellen (HSCs) (Yoshiji et al., 2001). Es wurde berichtet, dass MMP-9 die Infiltration von Neutrophilen und Makrophagen durch Abbau von interzellulären Matrizen wie Vitronektin und Fibronektin induziert, was zu einer Verstärkung der Entzündung führt (Medina et al., 2006). Bei NASH-Patienten wurde ein signifikanter Anstieg der MMP-9-Genexpression in der Leber im Vergleich zu normalen Kontrollen beobachtet (Ljumovic et al., 2004). Hepatische Überexpression von TGF-β in transgenen Mäusen führte zu schwerer Leberfibrose durch Augmentation der Prokollagen-I-Genexpression (Casini et al., 1993). Es ist bekannt, dass sowohl die TGF-β-Bildung als auch die Kollagen-I-Akkumulation eine Leberfibrose induzieren. Die Aktivierung von SCF induziert eine Erhöhung der Mastzellzahl, und seine enzymatische Funktion kann zu einer Erhöhung der Chymaseaktivität in fibrotischen Geweben führen (Maruichi et al., 2004). Diese enzymatischen Funktionen der Chymase können an Steatose, Entzündung und Fibrose beteiligt sein, die alle in der Leber von NASH-Patienten und Tiermodellen beobachtet werden (Abbildung 2).
Beteiligung von Chymase an NASH-Tiermodellen
Die Methionin- und Cholin-defiziente (MCD) Diät wurde weit verbreitet verwendet, um ein typisches NASH-Modell zu induzieren. Bei Hamstern, die mit der MCD-Diät gefüttert wurden, wurden signifikante Anstiege von Gesamtbilirubin, Triglycerid und Hyaluronsäure im Plasma beobachtet (Tashiro et al., 2010). Darüber hinaus wurden eine Akkumulation von Entzündungszellen und eine Zunahme der Lipidablagerungsfläche und der fibrotischen Fläche in der Leber beobachtet. In diesem MCD-Diät-induzierten NASH-Modell waren die hepatische Chymaseaktivität und verwandte Faktoren wie Angiotensin II, MMP-9 und Kollagen I signifikant erhöht (Tashiro et al., 2010; Masubuchi et al., 2013). Kürzlich wurde ein neues NASH-Modell entwickelt, bei dem schlaganfallanfällige spontan hypertensive 5 / Dmcr (SHRSP5 / Dmcr) Ratten mit einer fettreichen und cholesterinreichen (HFC) Diät gefüttert wurden (Kitamori et al., 2012). Dieses Modell zeigte Symptome des metabolischen Syndroms, von denen angenommen wurde, dass sie klinisch denen von NASH-Patienten ähneln (Kitamori et al., 2012). Im HFC-Diät-induzierten NASH-Modell wurden Hypertonie und Hyperlipidämie beobachtet und schwere Steatose, Fibrose und entzündliche Zellakkumulation in der Leber nachgewiesen (Miyaoka et al., 2017). Ferner wurde eine signifikante Zunahme der Chymaseaktivität zusammen mit MMP-9, TGF-β und Kollagen I in der Leber beobachtet (Miyaoka et al., 2017). So scheint es eine enge Beziehung zwischen Chymase und NASH Pathogenese in Tiermodellen von NASH zu geben.
Wirkung von Chymasehemmer in NASH-Tiermodellen
Wirkung von Chymasehemmer in NASH-Tiermodellen
Ein niedermolekularer Chymasehemmer schwächte die Chymaseaktivität signifikant ab und verringerte die Angiotensin-II-, MMP-9- und Kollagen-I-Spiegel in der Leber in einem mit MCD-Diät gefütterten NASH-Hamstermodell, wenn die Verabreichung des Inhibitors gleichzeitig mit der MCD-Diät begonnen wurde (Tashiro et al., 2010; Masubuchi et al., 2013). Der Chymasehemmer verhinderte in diesem NASH-Modell signifikant Lebersteatose, Fibrose und entzündliche Zellakkumulation (Tashiro et al., 2010; Masubuchi et al., 2013). Es wird angenommen, dass oxidativer Stress eine Rolle in der Multiple-Hit-Theorie der NASH-Entwicklung spielt, und die Augmentation des oxidativen Stressmarkers Malondialdehyd wurde in der Leber durch den Chymasehemmer signifikant abgeschwächt (Masubuchi et al., 2013). In einem Hamster-MCD-Diät-induzierten NASH-Modell zeigte der Chymase-Inhibitor eine Besserungswirkung, wenn er in etabliertem NASH verabreicht wurde (Masubuchi et al., 2013). Der Grad der Steatose und Fibrose in der Leber war im Vergleich zu vor der Verabreichung des Chymasehemmers reduziert (Masubuchi et al., 2013).
In der Leber eines hypertensiven Ratten-HFC-Diät-induzierten NASH-Modells dämpfte ein niedermolekularer Chymase-Inhibitor die Chymase-Spiegel sowie MMP-9, TGF-β und Kollagen I, die alle Chymase-assoziierte Faktoren sind (Miyaoka et al., 2017). Der Chymasehemmer schwächte die hepatische Steatose und Fibrose signifikant ab und reduzierte die Myeloperoxidase als Entzündungsmarker, insbesondere der Neutrophilen Infiltration (Miyaoka et al., 2017). In diesem HFC-Diät-induzierten Modell betrug das Überleben der mit Placebo behandelten Gruppe 14 Wochen nach Beginn der HFC-Diät 0% und resultierte aus schwerem Leberversagen (Miyaoka et al., 2017). Die mit Chymase-Inhibitor behandelte Gruppe, in der die Ratten unmittelbar nach Beginn der HFC-Diät mit dem Chymase-Inhibitor behandelt wurden, zeigte jedoch nach 14 Wochen ein 100% iges Überleben. Darüber hinaus wurde eine Überlebensrate von 50% für Ratten berichtet, die mit dem Chymase-Inhibitor behandelt wurden, beginnend 8 Wochen nach Beginn der HFC-Diätfütterung, zu welchem Zeitpunkt NASH etabliert wurde (Miyaoka et al., 2017).
Daher könnten Chymase-Inhibitoren nützliche Mittel zur Vorbeugung und Verbesserung von NASH in Tiermodellen sein. Andererseits fördert Angiotensin II indirekt auch Leberentzündungen, Steatose und Fibrose durch Erhöhung der MMP-9- und TGF-β-Genexpression. Sowohl MMP-9 als auch TGF-β sind eng an der Pathogenese von NASH beteiligt, aber diese Faktoren werden nicht notwendigerweise nur durch Angiotensin II induziert (Takai et al., 2010). Andere Faktoren als die Angiotensin-II-Stimulation tragen zum Anstieg der MMP-9- und TGF-β-Genexpression bei (Takai et al., 2010). In solchen Fällen ist der Angiotensin-II-Rezeptorblocker (ARB) nicht in der Lage, MMP-9- und TGF-β-Wirkungen abzuschwächen; Ein Chymasehemmer könnte jedoch abschwächende Wirkungen durch Hemmung der MMP-9- und TGF-β-Aktivierung haben, was auf einen möglichen Behandlungsverlauf zur Verhinderung der NASH-Progression hinweist.
Mechanismus der durch Chymasehemmer abgeschwächten Leberentzündung
Chymasehemmer konnte Entzündungen in Hamster-MCD-Diät- und Ratten-HFC-Diät-induzierten NASH-Modellen reduzieren (Tashiro et al., 2010; Masubuchi et al., 2013; Miyaoka et al., 2017). Die Behandlung mit Chymasehemmern schwächte die Chymaseaktivität in der Leber signifikant ab und reduzierte die Angiotensin-II- und MMP-9-Spiegel (Tashiro et al., 2010; Masubuchi et al., 2013; Miyaoka et al., 2017). In HSC induziert Angiotensin II die ROS-Erzeugung wie Wasserstoffperoxid und Superoxid durch die Aktivierung der Nicotinamidadenindinukleotidphosphat (NADPH) -Oxidase (De Minicis und Brenner, 2007). Chymase-Inhibitor führte zu einer Reduktion der Genexpression der NADPH-Oxidase-Komponente Rac-1 und des oxidativen Stressmarkers Malondialdehyd zusätzlich zu einer Reduktion der Angiotensin-II-Spiegel in einem Hamster-MCD-induzierten NASH-Modell (Masubuchi et al., 2013). Angiotensin II-induzierte Augmentation von ROS förderte die MMP-9-Genexpression in Neutrophilen und Makrophagen (Yaghooti et al., 2011; Kurihara et al., 2012). Daher hemmt der Chymasehemmer direkt die Aktivierung von proMMP-9 zu MMP-9 und reduziert indirekt die MMP-9-Genexpression über vermindertes Angiotensin II. MMP-9 spaltet extrazelluläre Matrixbestandteile wie Vitronektin und Fibronektin, führt zum Zerfall der Leberintegrität und induziert die Infiltration von Makrophagen und Neutrophilen (Medina et al., 2006). In einem HFC-Diät-induzierten NASH-Modell wurde ein signifikanter Anstieg der Myeloperoxidase-Expression in Makrophagen und Neutrophilen in der Leber beobachtet und durch Chymasehemmer reduziert (Miyaoka et al., 2017). Daher kann der durch den Chymasehemmer abgeschwächte Entzündungsmechanismus von der Verringerung der Angiotensin-II- und MMP-9-Spiegel in der Leber abhängen.
Mechanismus der durch Chymasehemmer abgeschwächten Lebersteatose
Angiotensin II kann die Lebersteatose über die ROS-Produktion beeinflussen. In muriner HSC verringerte ein Inhibitor der NADPH-Oxidase die ROS-Produktion signifikant und ein ARB verlangsamte die Entwicklung einer Lebersteatose durch Abschwächung der ROS-Produktion (Hirose et al., 2007; Guimarães et al., 2010). In einem MCD-Diät-induzierten NASH-Mausmodell wurde bei Angiotensin-II-Rezeptor-defizienten Mäusen eine signifikante Abschwächung der Steatose beobachtet (Nabeshima et al., 2009). Sowohl In-vivo- als auch In-vitro-Experimente zeigten, dass Angiotensin II die Genexpression des sterolregulatorischen Elementbindungsproteins (SREBP) -1c und der Fettsäuresynthase (FAS), die beide wichtige Faktoren bei der Regulation der Lipogenese sind, nach ROS-Augmentation hochregulierte (Kim et al., 2001; Hongo et al., 2009). Im Gegensatz dazu attenuierte ARB die hepatische Steatose zusammen mit der Herunterregulierung der Genexpression von SREBP-1c und FAS über die ROS-Attenuierung in einem Maus-NASH-Modell (Kato et al., 2012). In einem Hamster-MCD-Diät-induzierten NASH-Modell wurde nach Behandlung mit einem niedermolekularen Chymase-Inhibitor eine signifikante Abschwächung der SREBP-1c- und FAS-Genexpression beobachtet (Masubuchi et al., 2013). Daher kann der Verbesserungsmechanismus der Lebersteatose durch einen Chymasehemmer von der Verringerung der ROS-Produktion über eine verringerte Angiotensin-II-Generation in der Leber abhängig sein.
Mechanismus der durch Chymasehemmer abgeschwächten Leberfibrose
Chymase kann eng mit dem Fortschreiten der Gewebefibrose assoziiert sein, da sie zur Bildung von TGF-β aus dem nicht bioaktiven Vorläufer TGF-β beiträgt, und es ist bekannt, dass TGF-β das Wachstum von Fibroblasten stark induziert (Takai et al., 2003; Oyamada et al., 2011). Es ist bekannt, dass TGF-β eine zentrale Rolle beim Fortschreiten der Fibrose bei NASH-Patienten über aktiviertes HSC spielt (Williams et al., 2000). Die Hemmung der TGF-β-Funktion über Genexpression und Signalgebung führte in experimentellen Modellen zu einer verbesserten Leberfibrose (George et al., 1999; Arias et al., 2003). In einem Ratten-HFC-Diät-induzierten NASH-Modell führte die Abschwächung der Chymaseaktivität durch einen Chymasehemmer zu einer Verringerung des TGF-β-Spiegels und der fibrotischen Fläche in der Leber (Miyaoka et al., 2017). Somit kann die Reduktion von TGF-β durch Chymasehemmer zur Vorbeugung von Leberfibrose beitragen.
Angiotensin II kann auch an der Induktion von Leberfibrose beteiligt sein. Angiotensin II induziert die Kontraktion und Proliferation von HSC und induziert auch die Genexpression von TGF-β in Fibroblasten in vitro (Kagami et al., 1994; Bataller et al., 2000). Sowohl die TGF-β-Spiegel als auch der Grad der Kollagenakkumulation und fibrotischen Läsionen wurden durch Gallengangligatur bei Wildtyp-Mäusen beobachtet, diese waren jedoch bei Mäusen mit Angiotensin-II-Rezeptor-Mangel abgeschwächt (Yang et al., 2005). In einem Ratten-NASH-Modell attenuierte ARB auch die Leberfibrose über die Reduktion der TGF-β-Genexpression (Hirose et al., 2007). Es kann auch eine andere Beziehung zwischen Angiotensin II und Leberfibrose als die Angiotensin II-induzierte TGF-β-Genexpression bestehen. Bei Patienten mit chronischer Hepatitis C reduzierte ARB die Kollagengenexpression über die Rac-1-Genexpression (Colmenero et al., 2009). HSC sind als die wichtigsten produzierenden Zellen von Kollagen in der Leber anerkannt, und eine Zunahme der Expression von α-glattem Muskelaktin (SMA) in HSC induziert stark die Ablagerung extrazellulärer Matrix, einschließlich Kollagen I (De Minicis und Brenner, 2007). Angiotensin II kann die α-SMA-Genexpression in Ratten-HSC induzieren. Im Gegensatz dazu führt eine Angiotensin-II-Blockade zu einer Abschwächung der Leberfibrose zusammen mit einer Reduktion von α-SMA (Yoshiji et al., 2001). Obwohl bei Patienten mit NASH nicht untersucht, waren sowohl Chymase- als auch Angiotensin-II-bildende Aktivitäten in fibrotischen Regionen von Lebern von Patienten mit Zirrhose signifikant erhöht, und es wurden signifikante Korrelationen zwischen Chymase, Angiotensin-II-bildender Aktivität und Leberfibrose beobachtet (Komeda et al., 2008). In einem Hamstertetrachlorid-induzierten Leberzirrhose-Modell wurden signifikante Erhöhungen der Chymase- und Angiotensin-II-bildenden Aktivität beobachtet, die zusammen mit einer Leberzirrhose nach Behandlung mit einem niedermolekularen Chymase-Inhibitor signifikant abgeschwächt wurden (Komeda et al., 2010).
Der Mastzellstabilisator Tranilast könnte die Aktivierung von Mastzellen hemmen, die Freisetzung von Chymase blockieren und dadurch die Entwicklung von Leberfibrose in einem Ratten-Diabetes- und HFC-Diät-induzierten NASH-Modell verhindern (Uno et al., 2008). Chymase fördert die Proliferation von Mastzellen über SCF-Aktivierung durch seine enzymatische Funktion (Longley et al., 1997). In NASH-Tiermodellen reduzierte der Chymasehemmer den Anstieg der Mastzellzahl in der Leber, was zu einer verringerten Chymaseaktivität nach direkter Hemmung durch den Chymasehemmer und einer indirekten Verringerung der Chymaseexpression in Mastzellen führte (Masubuchi et al., 2013; Miyaoka et al., 2017).
Daher kann ein Chymasehemmer durch Hemmung der TGF-β-Aktivierung durch Chymasehemmung und / oder Abschwächung des TGF-β-Spiegels durch Verringerung von Angiotensin II und Mastzellproliferation zur Vorbeugung von Leberfibrose beitragen.
Schlussfolgerung
Das metabolische Syndrom, das Fettleibigkeit, Insulinresistenz, Hyperlipidämie und Hypertonie umfasst, steht in engem Zusammenhang mit der Entwicklung von NASH, und es wurden Studien mit Antidiabetika, Antihyperlipidämika und Antihypertensiva durchgeführt zur Behandlung von NASH. Das Konzept hinter diesen Mitteln besteht darin, die Symptome des metabolischen Syndroms abzuschwächen (Georgescu et al., 2009; Park et al., 2010; Mahady et al., 2011). Frühere Berichte haben gezeigt, dass Chymasehemmer Entzündungen und Fibrosen abschwächt, ohne den Blutzucker- und Lipidspiegel sowie den Blutdruck in Tiermodellen für Diabetes, Hyperlipidämie bzw., 2009; Takai et al., 2014; Zhang et al., 2016). Daher besteht das Konzept hinter der Chymase-Hemmung darin, Leberentzündungen und Fibrosen von NASH direkt abzuschwächen. Wir schlagen vor, dass der auf das metabolische Syndrom abzielende Chymasehemmer eine potenziell wirksame Strategie zur Abschwächung der NASH-Progression darstellt.
Autorenbeiträge
ST und DJ: schrieb das Manuskript. Beide Autoren lasen und genehmigten das endgültige Manuskript.
Interessenkonflikterklärung
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.