Einleitung
Die Limb-Girdle Muscular Dystrophies (LGMDs) umfassen eine heterogene Gruppe von Erkrankungen, die durch fortschreitende Verschwendung und Schwäche der Muskeln des proximalen Gliedgürtels gekennzeichnet sind (1, 2). LGMDs zeigen inter- und intrafamiliale Variabilität, die von sehr milden Formen bis zu schweren, früh einsetzenden, schnell fortschreitenden Phänotypen reicht (2). LGMDs können als autosomal dominant (LGMD1) oder rezessiv (LGMD2) klassifiziert werden. Die erste Gruppe hat normalerweise ein erwachsenes Erkrankungsalter und ist nicht sehr häufig (< 10% aller LGMDs), während letztere häufiger auftreten (1:15000) (1). Unter den rezessiven Formen ist die LGMD2A (oder Calpainopathie) die häufigste LGMD weltweit und betrifft etwa 30% aller LGMDs-Fälle (3). Klinische Signaturen der Krankheit sind Zehenspitzen gehen, Watscheln Gang, Schwierigkeiten beim Laufen und Treppensteigen, Skapulierflügel. Gelenkkontrakturen, Achillessehnenverkürzungen und Skoliosen werden häufig beobachtet, während Gesichts- und Nackenmuskeln nicht betroffen sind (4). Eine asymptomatische Hyperglykämie (5-80-fache CPK-Normalwerte) bei jungen Patienten gilt als präklinisches Krankheitsstadium und kann mehrere Jahre andauern (1, 5). Das Alter des Beginns der Muskelschwäche tritt normalerweise im Alter von 15 Jahren auf, obwohl es im Alter von früher (< 12 Jahre) oder später (> 30 Jahre) auftreten kann. Das Fortschreiten der Krankheit kann im fortgeschrittenen Stadium zu Gehbehinderung, Ateminsuffizienz und verminderter Lungenvitalkapazität führen (6). Eine Herzbeteiligung (Herzrhythmusstörungen, Herzleitungsstörungen, linksventrikuläre Ejektionsstörung) wird nur gelegentlich berichtet (6, 7).
Die Diagnose einer Calpainopathie wird durch den Nachweis pathogener Mutationen in CAPN3 (15q15.1) (4) bestätigt, die für verschiedene alternativ gespleißte Transkripte kodieren. Das Transkript in voller Länge wird jedoch hauptsächlich im Muskelgewebe exprimiert (8). Das codierte Protein (CAPN3) gehört zur Familie der nicht-lysosomalen Ca ++ -abhängigen Cysteinproteasen. Im Muskel nimmt CAPN3 am „Sarkomerumbau“ teil, der für die Muskelanpassung und das Muskelwachstum als Reaktion auf funktionelle und metabolische Anforderungen unerlässlich ist. Bis heute wurden mehr als 490 pathogene Mutationen in CAPN3 beschrieben, von denen die meisten Einzelnukleotidveränderungen sind (4). Mutationen in CAPN3 wurden mit mitochondrialen Anomalien, Wachstumsversagen, erhöhtem oxidativem Stress und Sarkomer-Desorganisation in Verbindung gebracht, die zusammen dazu beitragen, dass der Muskel nicht in der Lage ist, Lasten zu halten, wodurch Myofaserdegeneration und Muskelschwund verursacht werden (8). Die Diagnose einer Calpainopathie kann aufgrund der genetischen Heterogenität und der Unspezifität des klinischen und instrumentellen Musters eine Herausforderung darstellen. In der Tat werden Symptome von Muskelschwäche / -atrophie, Hypertrophie / Pseudohypertrophie und Sehnenkontrakturen sehr häufig mit anderen LGMDs oder neuromuskulären Störungen geteilt. Das Muskelbiopsiemuster bei der Calpainopathie ist im Allgemeinen ebenfalls unspezifisch und reicht von leichten Muskelanomalien bis zu schweren dystrophischen Veränderungen. Darüber hinaus sind immunhistochemische / biochemische Marker normalerweise nicht zuverlässig: das Calpain-Signal kann auch bei Vorhandensein eines nicht funktionellen Proteins normal sein und umgekehrt kann es auch bei anderen Muskeldystrophien, die sich von der Calpainopathie unterscheiden, reduziert sein.
Es wird daher empfohlen, eine Differentialdiagnose durchzuführen, um genaue und zuverlässige Ergebnisse zu liefern. Zu diesem Zweck wurden molekulargenetische Testansätze entwickelt, um die Diagnose der Calpainopathie zu bestätigen, einschließlich Multigenpanel, umfangreiche genomische (Exom- / Genomsequenzierung) und Einzelgenanalyse (direkte Sequenzierung) (9). Die Implementierung von Next-Generation Sequencing (NGS) war hilfreich, um aussagekräftige Daten für diagnostische, prädiktive oder therapeutische Zwecke zu generieren (10-12). NGS-Genpanels basieren auf der Analyse einer Reihe von Genen, die mit einer bestimmten Krankheit oder einer Gruppe verwandter Störungen assoziiert sind, die durch genetische und phänotypische Heterogenität gekennzeichnet sind. NGS-Panels können auch nützlich sein, um gemischte oder komplexe Phänotypen zu erkennen, die aus der Vererbung von mehr als einem genetischen Defekt von den Eltern resultieren (13). Im Allgemeinen sind Patienten mit komplexen Phänotypen, die negativ auf die bekannten Mutationen in maßgeschneiderten diagnostischen Genpanels reagieren und eine breite Differentialdiagnose erfordern, für die gesamte Exom- oder Genomsequenzierung geeignet. Die Sequenzierung des gesamten Exoms / Genoms ist ein teurerer Ansatz in Bezug auf Datenmanagement, Interpretation und Analysekosten, erhöht jedoch die Wahrscheinlichkeit, eine molekulare Diagnose einer vermuteten genetischen Störung zu stellen (13). In Bezug auf LGMDs werden dedizierte NGS-Panels dringend empfohlen, um hohe Diagnoseraten, optimale Abdeckung, Sensitivität und Spezifität klinischer Tests zu gewährleisten (9). NGS-Panels stellen daher eines der besten Systeme dar, um die Differentialdiagnose zu erleichtern, neue ursächliche Mutationen zu identifizieren und Genotyp-Phänotyp-Korrelationen zu klären (14, 15).
In diesem Manuskript wird der Fall eines von LGMD2A betroffenen Patienten berichtet, bei dem durch die Anwendung des NGS-Panels nicht nur die Diagnose einer Calpainopathie (Mutationen in CAPN3) bestätigt, sondern auch eine zusätzliche, neuartige Mutation im LMNA-Gen identifiziert werden konnte, die mit einer dilatativen Kardiomyopathie assoziiert ist. Angesichts dieser Ergebnisse wurde die Analyse auf die Familienmitglieder des Probanden ausgeweitet, um eine umfassendere Interpretation der Analysedaten in Bezug auf die pathologischen Phänotypen zu ermöglichen.
Fallpräsentation
Klinische Charakterisierung von Probanden und Verwandten
Der Ausbruch der Krankheit bei der Probandin wurde im Alter von 10 Jahren gemeldet, als sie über das Vorhandensein einer Wadenhypertrophie mit Zehenspitzen, Schwierigkeiten beim Laufen, Treppensteigen und Stehen berichtete. Die Symptome zeigten eine langsame, aber fortschreitende Verschlechterung mit anschließender Beteiligung der proximalen oberen Extremität. Im Alter von 13 Jahren wurden übrigens hohe CPK-Spiegel (~ 8.000 UI / L) entdeckt, die niemals mit Myoglobinurie assoziiert waren. Nacheinander unterzog sich der Patient mehreren neuromuskulären Untersuchungen in verschiedenen spezialisierten Zentren. Es wurden zwei Muskelbiopsien durchgeführt, die ein klassisches dystrophisches Bild (hypertrophe / atrophische Fasern, innere Kerne, nekrotische Fasern, Zunahme des Bindegewebes) mit unspezifischen Eigenschaften zeigten. Wie erwartet waren Immunochemie- und Immunoblot-Studien nicht schlüssig, was auf ein abnormales / reduziertes α-, β-, γ-Sarkoglycan-, β-Dystroglycan- und Dystrophin-Signal nur in nekrotischen Fasern hinwies (wiederholte Immunochemie ergab normal, auch für Laminin); Immunoblots für Dystrophin und Calpain zeigten normale Signale. Es ist jedoch bekannt, dass der Calpain-3-Immunoblot für die LGMD2A-Diagnose nicht vollständig empfindlich ist, da 20-30% der Fälle eine normale Proteinmenge aufweisen (1, 16-18). Aufgrund des klinischen Erscheinungsbildes (Hyperrhythmie, Wadenhypertrophie/Pseudohypertrophie) wurden Dystrophinopathien zunächst ausgeschlossen. Darüber hinaus ergab die Analyse von DMD (Xp21) -, FKRP (19q13.32) – und DYSF (2p13.2) -Genen keine pathogenen Mutationen. Der Patient kam im Alter von 30 Jahren zu unserer Beobachtung und zeigte eine axiale und Gürtelbeteiligung sowohl in der oberen als auch in der unteren Extremität mit signifikantem watschelndem Gang und geflügelten Schulterblättern. In den unteren Extremitäten war die Schwäche in den Quadrizeps- und Gesäßmuskeln, die signifikant pseudoatrophisch waren, zusammen mit dem Hinweis auf „scheinbar hypertrophe“ (Pseudo-Hypertrophie) Wadenmuskeln (Abbildung 1). Krämpfe, Myalgien und Kräuselungen wurden nicht beobachtet. Vollständige pneumologische (Spirometrie und Polysomnographie) und kardiologische (Echographie, 24-h-EKG-Holter-Monitoring, Herz-MRT, vollständige kardiologische Untersuchung mit gezielter Anamnese und kardiovaskulärer Reflexanalyse) Untersuchungen ergaben keine signifikanten Anomalien. Wir schlossen auch einen Mangel an saurer Maltase aus (normale Enzymaktivitätstests in Lymphozyten / Leukozyten). Die Magnetresonanztomographie (MRT) der Muskeln zeigte eine ausgeprägte Beteiligung des Schultergürtels, der paravertebralen Muskeln, der hinteren Kompartimentsmuskeln des Oberschenkels (mit relativer Schonung der Muskeln Sartorius und Gracilis) und des Beins (insbesondere Gastrocnemius medialis und Soleus) (Abbildung 2). Dieses Beteiligungsmuster wurde bereits in der Literatur zum MRT-Bild bei der Calpainopathie berichtet (19, 20). Darüber hinaus zeigte die MRT-Studie, dass Wadenmuskeln effektiv atrophisch waren und durch Fettinfiltration gekennzeichnet waren, was zu einer muskulären „Vergrößerung“ führte.“ Das klinische Erscheinungsbild, das Erkrankungsalter, die Progressionsrate, die Verteilung der Muskelschwäche und die MRT-Befunde im Probanden stimmten vollständig mit einer in der Literatur ausführlich beschriebenen Diagnose von LGMD2A überein (4, 21, 22).
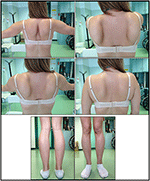
Abbildung 1. Klinische Merkmale des Probanden: geflügelte Schulterblätter und Kombination von Oberschenkelatrophie und Wadenpseudohypertrophie.
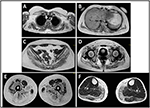
Abbildung 2. Proband Muskel MRT: beteiligung des oberen Extremitätengürtels und der paravertebralen Muskulatur (A,B), Beteiligung des unteren Extremitätengürtels und des hinteren Oberschenkelkompartiments (C–E, meist Glutei) mit relativer Schonung von Sartorius und Gracilis und Beteiligung des hinteren Beinkompartiments (F, meist Gastrocnemius medialis / soleus).
Die Auswertung der Familienmitglieder des Probanden ergab keine Vorgeschichte neuromuskulärer Erkrankungen und keine Hinweise auf Blutsverwandtschaft zwischen den Eltern. Die Mutter des Probanden präsentierte jedoch im Alter von 55 Jahren 2 synkopale Episoden, bei denen idiopathische Bradykardie diagnostiziert wurde (bis zu 40 Schläge pro Minute bei Nacht). Umfangreiche kardiologische Untersuchungen an der Mutter des Probanden diagnostizierten nach Auftreten mehrerer synkopaler Episoden/Lipothymie einen symptomatischen atrioventrikulären (AV) Block. Die Analyse zeigte das Vorhandensein eines grundlegenden AV-Blocks ersten Grades mit mehreren Perioden schwerer Bradykardie, meist nächtlich und oft symptomatisch, verbunden mit einigen Phasen eines AV-Blocks zweiten Grades, sowohl vom Typ 1 als auch vom Typ 2, und einigen Phasen einer nächtlichen vollständigen AV-Dissoziation. Echokardiographie und ergometrischer Test zeigten keine signifikanten Veränderungen, mit Ausnahme des Foramen ovale des Patents. Die Mutter zeigte kein anderes klinisches Symptom, keinen prädisponierenden Zustand oder Risikofaktor. Angesichts ihres klinischen Bildes wurde der Mutter des Probanden PMK implantiert. Darüber hinaus ergab die Familiengeschichte, dass der Großmutter und dem Urgroßvater des Probanden im Alter von 55 bzw. 30 Jahren ebenfalls PMK implantiert wurde. Darüber hinaus starb der Bruder der Großmutter des Probanden im Alter von 51 Jahren an schwerer dilatativer Kardiomyopathie. Kardiologische Untersuchungen wurden auch am Vater und am Onkel mütterlicherseits des Probanden durchgeführt, die völlig unberührt blieben.
Die vorliegende Studie wurde von der Ethikkommission der Santa Lucia Foundation genehmigt und gemäß der Erklärung von Helsinki durchgeführt. Alle Teilnehmer gaben eine unterzeichnete Einverständniserklärung zur genetischen Analyse ab, und in diesem Zusammenhang gaben sie auch die Zustimmung zur Veröffentlichung dieses Fallberichts.
Laboruntersuchungen und diagnostische Tests
Genomische DNA wurde aus peripherem Blut (400 µL) mit dem MagPurix Blood DNA Extraction Kit und dem MagPurix Automatic Extraction System (Resnova) gemäß den Anweisungen des Herstellers extrahiert. Die Proben wurden mit dem Ion PGM System und dem Ion Ampliseq Customized Panel High Specificity (Thermo Fisher Scientific) sequenziert. Die Größe des Panels betrug 129.13 Kb, was voraussichtlich ~ 99,72% des gesamten Panels mit einer Mindestabdeckung von 20X screenen wird. Das Panel umfasste 18 Gene, die anhand wissenschaftlicher Literatur, GeneReviews (www.ncbi.nlm.nih.gov/books/NBK1116 /) und Häufigkeit pathogener Varianten in der Allgemeinbevölkerung. Eine detaillierte Beschreibung des NGS-Panels ist in Tabelle 1 zusammengefasst.
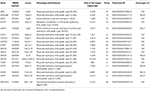
Tabelle 1. Angepasst NGS panel verwendet für LGMD diagnose.
Bibliotheken Der Aufbau wurde mit Ion AmpliSeq™ Library Kits 2.0 durchgeführt. Für Multiplex-PCR-Reaktionen wurden ca.10 ng/µl Ausgangs-DNA verwendet. Nacheinander wurden zwei Reinigungsschritte (unter Verwendung von AMPure XP, Beckman Coulter) durchgeführt, um unerwünschte Verunreinigungen zu entfernen, und eine abschließende PCR wurde durchgeführt. Die Template-Amplifikations- und Anreicherungsschritte wurden mit Ion PGM Hi-Q OT2 kit-400, Ion OneTouch 2 System und Ion OneTouch ES (Thermo Fisher Scientific) durchgeführt. Die Proben wurden mit dem Ion PGM Hi-Q Sequencing Kit (400 bp, Thermo Fisher Scientific) verarbeitet und auf dem Ion 316 Chip v2 (850 bp erforderlich) und dem Ion PGM Sequencer (Thermo Fisher Scientific) ausgeführt. Die Ergebnisse wurden mit Ion Reporter 4.6 (Thermo Fisher Scientific) und Integrated Genome Viewer (IGV) analysiert. Die Interpretation genetischer Varianten wurde von der Human Gene Mutation Database (HGMD), der Leiden Open Variation Database (LOVD), ClinVar und ExAC durchgeführt. Die funktionelle Wirkung der nachgewiesenen Varianten wurde mit bioinformatischen Vorhersagewerkzeugen bewertet, darunter Mutation Taster, Varsome, SIFT, Polyphenol, SMART, Human Splicing Finder (HSF). Direkte Sequenzierung (BigDye Terminator v3.1, BigDyeX Terminator und ABI3130, Applied Biosystems) wurde durchgeführt, um genetische Varianten zu bestätigen und genomische kodierende Regionen mit einer Abdeckung <20X zu sequenzieren (LMNA, Chr1:156106052-156106076; DYSF, Chr2: 71753352-71753502, Chr2: 71776385-71776640; SGCB, Chr4: 52904225-52904560; SGCA, Chr17:48243242-48243570).
Ergebnisse
Die NGS-Analyse des Probanden ergab 3 heterozygote Varianten (Ergänzende Abbildung 1). In CAPN3 wurden zwei Varianten lokalisiert, nämlich NM_000070.2 (CAPN3): c.550delA (p.Thr184Argfs) und c.1813G> C (p.Val605Leu) in den Exons 4 bzw. 16. Die erste ist eine einzelne Nukleotiddeletion und ist die häufigste (75% der Fälle) pathogene Variante in europäischen Ländern (1). Erwartungsgemäß stufte die bioinformatische Analyse den c.550delA (rs80338800) als eine Loss-of-Function-Variante (p.Thr184Argfs) ein, was zu einer Frameshift des offenen Leserahmens führte. Die Vorhersagewerkzeuge (Mutation Taster, HSF, Varsome, Polyphenol, SIFT) beschrieben die Variante als schädlich. Darüber hinaus zeigte SMART, dass dem veränderten Proteinprodukt die Calpain 3-Domäne und die drei „EF-Hands“ -Motive fehlen. Die erste Domäne ist an den Signalwegen von Calpain beteiligt, während die EF-Hände für die Ca ++ -abhängige Aktivierung des Proteins essentiell sind (4).
In Bezug auf c.1813G> C handelt es sich um eine neuartige Missense-Variante (p.Val605Leu), von der vorhergesagt wurde, dass sie schädlich ist (durch Mutation Taster, HSF, Varsome, Polyphenol, SIFT) wegen einer möglichen Veränderung des Spleißens. Diese Variante ist in der Literatur oder in Online-Datenbanken nicht annotiert und wurde bei 200 Kontrollpersonen nicht gefunden. Leider ergab die Analyse durch SMART Tool keine signifikanten Ergebnisse, da sich die Variante in einer nicht charakterisierten Domäne befindet. Die Segregationsanalyse von CAPN3 zeigte, dass die Mutter und der Onkel mütterlicherseits beide heterozygot für c.550delA waren, während der Vater Träger von c.1813G> C.
Darüber hinaus ergab die NGS-Analyse des Probanden das Vorhandensein einer neuartigen Variante in LMNA, nämlich NM_170707 (LMNA): c.550C>T (p.Gln184 *). Die oben erwähnte Variante wurde als Nullvariante vorhergesagt, wurde noch nicht in der Literatur oder in Online-Datenbanken beschrieben und bei 200 Kontrollpersonen nicht gefunden. Prädiktive Werkzeuge (Mutation Taster, HSF, Varsome, Polyphenol, SIFT) beschrieben eine pathogene Wirkung dieser Variante auf das Proteinprodukt. SMART Tool berichtete, dass dem veränderten LMNA-Protein die Filamentdomäne fehlt, die für die Aufrechterhaltung seiner Struktur und Funktion unerlässlich ist. Die Segregationsanalyse zeigte das Vorhandensein des c.550C > T-Variante nur bei der Mutter, was auf einen möglichen Zusammenhang mit ihrer kardialen Symptomatik und ihrer Familienanamnese hindeutet.
Nach den Kriterien der Standards und Richtlinien des American College of Medical Genetics (ACMG) (23) kann c.1813G>C (CAPN3) als wahrscheinlich pathogene Variante beschrieben werden, wenn man bedenkt, dass sie sich in einer kritischen Domäne ohne gutartige Variation befindet (PM1); Es fehlt in ExAc-, GnomAD- und 1000 Genome Browser-Datenbanken (PM2); in Anbetracht des rezessiven Vererbungsmodells wurde es bei trans mit einer pathogenen Variante (PM3) nachgewiesen; Mehrere Zeilen von Computerbeweisen unterstützen eine schädliche Wirkung auf das Gen oder Genprodukt (PP3). In Bezug auf die klinische Klassifizierung von c.550C > T (LMNA) durch ACMG kann es als pathogene Variante bezeichnet werden, da es sich um eine Nullvariante handelt, die in LMNA (PVS1) zu Funktionsverlust führt (p.Gln184 *); es fehlt in ExAc-, GnomAD- und 1000 Genome Browser-Datenbanken (PM2); Es befindet sich in einer kritischen Domäne ohne gutartige Variation (PM1); mehrere rechnerische Evidenzlinien unterstützen eine schädliche Wirkung auf das Gen oder Genprodukt (PP3).
Schlussfolgerungen
In diesem Fallbericht wurde ein von LGMD betroffener Patient vorgestellt, der nicht nur in CAPN3 (c.550delA und c.1813G>C), sondern auch in LMNA (c.550C>T) Mutationsträger war. Diese Ergebnisse erklären den neuromuskulären Phänotyp des Probanden aufgrund von CAPN3-Mutationen und heben ein potenzielles Risiko für Herz-Kreislauf-Erkrankungen aufgrund des Vorhandenseins der Variante in LMNA und ihrer positiven Familienanamnese hervor. Angesichts dieser Ergebnisse wurden die Familienmitglieder der Segregationsanalyse unterzogen. Insbesondere ergab sich, dass die Mutter Träger von CAPN3_c.550delA bzw. LMNA_c.550C>T war. Die Mutter kann als gesunde Trägerin der mit Calpainopathie assoziierten pathogenen Mutation CAPN3_c.550delA angesehen werden. Das Vorhandensein einer neuartigen Variante in LMNA kann jedoch ihre kardiovaskuläre Pathologie (Bradykardie und synkopale Episoden) erklären. Das Fehlen einer neuromuskulären Symptomatik bei der Mutter und das eigentümliche Krankheitsbild des Probanden schlossen eine mögliche Assoziation von LMNA_c aus.550C>T mit neuromuskulärem Phänotyp, insbesondere bezüglich Emery-Dreifuss-Muskeldystrophie (EMD). Tatsächlich betrifft EMD speziell die Muskeln Vastus lateralis und Bizeps brachii, die bei LGM2A relativ verschont bleiben (24, 25).
Der Onkel mütterlicherseits war nur für die CAPN3_c.550delA heterozygot und zeigte erwartungsgemäß keine neuromuskulären oder kardiovaskulären Probleme. Der Vater trug weiterhin den Roman CAPN3_c.1813G>C und war dadurch nicht betroffen. Insgesamt bestätigte die Segregationsanalyse die Vererbung der drei Mutationen des Probanden von ihren Verwandten und hob eine Vertrautheit für die Kardiomyopathie hervor, die nicht vernachlässigt werden kann (Abbildung 3).
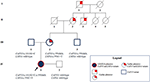
Abbildung 3. Stammbaum, der die positive Vertrautheit für den kardialen Phänotyp zeigt, der von der mütterlichen Abstammung geerbt wurde, und die Übertragung der CAPN3- und LMNA-Varianten durch die Familienmitglieder.
Insgesamt werfen diese Daten einige wichtige Überlegungen auf. Erstens war das NGS-Panel bei diesem Patienten entscheidend für die molekulare Diagnose der Calpainopathie, indem eine bekannte Mutation und eine zweite neuartige Variante als pathogen vorhergesagt wurden. Zweitens ermöglichte das NGS-Panel die Identifizierung des Romans LMNA_c.550C> T Variante im Probanden. Dieses Ergebnis stimmte mit der positiven Familienanamnese und den Segregationsdaten überein, erklärte Herz-Kreislauf-Erkrankungen bei der Mutter und, was noch wichtiger ist, empfahl eine spezifischere kardiologische Nachsorge beim Probanden. Es ist wichtig anzumerken, dass kardiologische Manifestationen bei der Mutter später im Alter (55 Jahre) auftraten, so dass wir annehmen können, dass sich der Proband noch in einer „asymptomatischen kardiologischen Phase“ befinden könnte.“ Eine variable phänotypische Expression der LMNA-Mutation im Probanden im Vergleich zur Mutter kann jedoch nicht ausgeschlossen werden. All diese Daten unterstreichen die Bedeutung eines integrierten Ansatzes zwischen Klinikern und Genetikern für die korrekte Interpretation der Ergebnisse, die ordnungsgemäße genetische Beratung und schließlich das klinische Management und Follow-up. In Bezug auf die genetische und familiäre Beratung wurde die NGS-Analyse auch am Partner des Probanden durchgeführt, um das reproduktive Risiko für das Paar abzuschätzen. Der Partner war für alle 18 getesteten Gene negativ, was bedeutet, dass er ein Restrisiko von 1/650 hat, um ein gesunder Träger für LGMD-ursächliche Mutationen zu sein, wenn man bedenkt, dass der Test zu 84% empfindlich ist. Unter Berücksichtigung des genetischen Profils des Probanden und der Sensitivität des NGS-Tests beträgt das Restrisiko für das Paar, ein von Calpainopathie betroffenes Kind zu bekommen, 1/1300. Andererseits werden die LMNA-pathogenen Varianten autosomal dominant übertragen, was bedeutet, dass der Proband eine Wahrscheinlichkeit von 50% hat, heterozygote Kinder zu haben. Das klinische Bild der Nachkommen kann jedoch nicht sicher vorhergesagt werden, da es sich bei LMNA_c.550C>T um eine neuartige Variante handelt und deren funktionelle Auswirkung auf den Phänotyp noch unbekannt ist.
Abschließend hebt dieser Fallbericht den klinischen Nutzen von NGS-Panels hervor, um eine genaue LGMD2A-Diagnose bereitzustellen und komplexe Phänotypen und Komorbiditäten zu beschreiben, die aus der Vererbung verschiedener Mutationen in mehreren Genen stammen. Die Anwendung von NGS in der klinischen Praxis sollte jedoch immer mit einer prä- und postgenetischen Beratung kombiniert werden, um eine klare Erklärung der Ergebnisse, der möglichen Auswirkungen auf den Phänotyp der Patienten, des Rezidivrisikos innerhalb der Familie sowie möglicher unerwarteter Befunde zu liefern.
Datenverfügbarkeit
Für diese Studie wurden keine Datensätze generiert oder analysiert.
Ethikerklärung
Diese Studie wurde in Übereinstimmung mit den Empfehlungen der Ethikkommission der Santa Lucia Foundation mit schriftlicher Einverständniserklärung aller Probanden durchgeführt. Alle Probanden gaben eine schriftliche Einverständniserklärung gemäß der Erklärung von Helsinki ab. Das Protokoll wurde von der Ethikkommission der Santa Lucia Foundation genehmigt.
Autorenbeiträge
RC, CSt, VC, GC, RG, GPa, SC, CP und JM leisteten Beiträge zur Datenerfassung, Analyse und Interpretation von Daten. CSt, RC, GC, RG, GM und EG waren an der Ausarbeitung des Manuskripts beteiligt. SZ, GPr, CSa und SS waren an der Erfassung klinischer Daten beteiligt. RC, CSt, VC, GC, RG, GPa, SC, CP, JM, SZ, GPr, GM, CSa, SS und EG haben die endgültige Genehmigung der zu veröffentlichenden Version erteilt.
Finanzierung
Diese Arbeit wird von 5X 2016 National Health Ministry 2 unterstützt.
Interessenkonflikterklärung
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Ergänzungsmaterial
Das Ergänzungsmaterial zu diesem Artikel finden Sie online unter: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. Protein und genetische Diagnose der Extremitätengürtel Muskeldystrophie Typ 2A: die Ausbeute und die Fallstricke. Muskelnerv. (2015) 52:163–73. doi: 10.1002/mus.24682
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
2. Nigro V, Savarese M. Genetische Grundlage von Muskeldystrophien des Gliedmaßengürtels: das Update 2014. Acta Myol. (2014) 33:1–12.
PubMed Zusammenfassung / Google Scholar
3. Richard I, Hogrel JY, Stockholm D, Payan CAM, Fougerousse F, Eymard B, et al. Naturgeschichte von LGMD2A zur Abgrenzung von Ergebnismessungen in klinischen Studien. In: Ann Clin Transl Neurol. (2016) 3:248–65. doi: 10.1002/acn3.287
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
4. Angelini C, Fanin M. Calpainopathie. In: Adam MP, Ardinger HH, Pagon RA Herausgeber. GeneReviews®. Seattle, WA: Universität von Washington (2017) 1-30.
PubMed Zusammenfassung / Google Scholar
5. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar in: Kyriakides T, Angelini C, Sacconi S, Siciliano G, Vilchez JJ, et al. EFNS-Richtlinien zum diagnostischen Ansatz bei pauci- oder asymptomatischer Hyperckämie. In: Eur J Neurol. (2010) 17:767–73. doi: 10.1111/j.1468-1331.2010.03012.x
CrossRef Volltext / Google Scholar
6. Mori-Yoshimura M, Segawa K, Minami N, Oya Y, Komaki H, Nonaka I, et al. Kardiopulmonale Dysfunktion bei Patienten mit Extremitätengürtel-Muskeldystrophie 2A. (2017) 55:465–9. doi: 10.1002/mus.25369
Querverweis Volltext / Google Scholar
7. Okere A, Reddy SS, Gupta S, Shinnar M. Eine Kardiomyopathie bei einem Patienten mit Extremitätengürtel-Muskeldystrophie Typ 2A. (2013) 6:e12–13. doi: 10.1161/CIRCHEARTFAILURE.112.971424
CrossRef Volltext / Google Scholar
8. Kramerova I., Ermolova N., Eskin A., Hevener A., Quehenberger O., Armando A.M., et al. Das Versagen, die Transkription von Genen, die für die Muskelanpassung notwendig sind, hochregulieren zu können, liegt der Muskeldystrophie des Gliedmaßengürtels 2A (Calpainopathie) zugrunde. Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093/hmg/ddw086
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
9. Thompson R, Straub V. Muskeldystrophien des Gliedmaßengürtels – internationale Kooperationen für translationale Forschung. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038/nrneurol.2016.35
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
10. Straella C, Caputo V, Galota MR, Zampatti S, Marella G, Mauriello S, et al. Anwendung der Präzisionsmedizin bei neurodegenerativen Erkrankungen. Front Neurol. (2018) 9:701. doi: 10.3389/fneur.2018.00701
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
11. Cascella R, Strafella C, Caputo V, Errichiello V, Zampatti S, Milano F, et al. Auf dem Weg zur Anwendung der Präzisionsmedizin bei altersbedingter Makuladegeneration. Prog Retin Auge Res. (2017) 63: 132-46. geburtsdatum: 10.1016/j.preteyeres.2017.11.004
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
12. Cascella R., Strafella C., Longo G., Manzo L., Ragazzo M., De Felici C., et al. Beurteilung des individuellen AMD-Risikos mit genetischer Beratung, Familienanamnese und Gentests. Auge. (2017) 32:446–50. doi: 10.1038/Auge.2017.192
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
13. Adams DR, Eng CM. Sequenzierung der nächsten Generation zur Diagnose vermuteter genetischer Störungen. In: N Engl J Med. (2019) 379:1353–62. doi: 10.1056/NEJMc1814955
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
14. Angelini C. Neuromuskuläre Krankheit. Diagnose und Entdeckung bei Extremitätengürtel-Muskeldystrophie. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038/nrneurol.2015.230
CrossRef Volltext / Google Scholar
15. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, et al. Verwendung der Whole-Exome-Sequenzierung zur Diagnose von Muskeldystrophie des Gliedmaßengürtels: Ergebnisse und Erkenntnisse. In: JAMA Neurol. (2015) 72:1424–32. ursprungsbezeichnung: 10.1001/jamaneurol.2015.2274
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
16. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar in: Milic A, Lochmüller H, Mora M, Moggio M, et al. Ein Drittel der LGMD2A-Biopsien weist eine normale proteolytische Aktivität von Calpain 3 auf, die durch einen In-vitro-Assay bestimmt wurde. In: Neuromusc Disord. (2007) 17:148–56. doi: 10.1016/j.nmd.2006.11.001
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
17. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar in: Lanzillo R, Aurino S, Fanin M, Aguennoz M, Vitale F, Fiorillo C, et al. Früh einsetzende Calpainopathie mit normalem, nicht funktionellem Calpain 3-Niveau. In: Dev Med Child Neurol. (2006) 48:304–6. doi: 10.1017/S001216220600065X
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
18. Talim B, Ognibene A, Mattioli E, Richard I, Anderson LV, Merlini L. Normale Calpain-Expression bei genetisch bestätigter Extremitätengürtel-Muskeldystrophie Typ 2A. Neurologie. (2001) 56:692–3. Ursprungsbezeichnung: 10.1212/wnl.56.5.692-a
CrossRef Volltext / Google Scholar
19. Díaz-Manera J, Llauger J, Gallardo E, Illa I. Muskel-MRT bei Muskeldystrophien. Acta Myol. (2015) 34:95–108.
PubMed Zusammenfassung / Google Scholar
20. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar in: Mercuri E, Bushby K, Ricci E, Birchall DR, Pane M, Kinali M, et al. Muskel-MRT-Befunde bei Patienten mit Extremitätengürtel-Muskeldystrophie mit Calpain-3-Mangel (LGMD2A) und frühen Kontrakturen. In: Neuromusc Disord. (2005) 15:164–71. doi: 10.1016/j.nmd.2004.10.008
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
21. Magri F, Nigro V, Angelini C, Mongini T, Mora M, Moroni ich, et al. Das italienische Register für Muskeldystrophie des Gliedmaßengürtels: relative Häufigkeit, klinische Merkmale und Differentialdiagnose. Muskelnerv. (2017) 55:55–68. doi: 10.1002/mus.25192
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
22. J. J. Chae, N. Minami, Y. Jin, M. Nakagawa, K. Murayama, F. Igarashi, et al. Calpain 3-Genmutationen: genetische und klinisch-pathologische Befunde bei Extremitätengürtel-Muskeldystrophie. In: Neuromusc Disord. (2001) 11:547–55. modell: 10.1016/S0960-8966(01)00197-3
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
23. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar in: Richards S, Aziz N, Bale S, Bick D, Gastier-Foster J, et al. Standards und Richtlinien für die Interpretation von Sequenzvarianten: eine gemeinsame Konsensempfehlung des American College of Medical Genetics and Genomics und der Association for Molecular Pathology. In: Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
24. Bonne G, Mercuri E, Muchir A, Urtizberea A, Bécane HM, Recan D, et al. Klinisches und molekulargenetisches Spektrum der autosomal dominanten Emery-Dreifuss-Muskeldystrophie aufgrund von Mutationen des Lamin A / C-Gens. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170:: AID-ANA6>3.0.CO ;2-J
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
25. J.H. Hong, J.S. Ki, J.W. Suh, J.L. Kim, K.K. Baek, et al. Herzrhythmusstörungen, Kardiomyopathie und Muskeldystrophie bei Patienten mit Emery-Dreifuss-Muskeldystrophie und Extremitätengürtel-Muskeldystrophie Typ 1B. J. Med Sci. (2005) 20:283–90. geburtsdatum: 10.3346/jkms.2005.20.2.283
Querverweis Volltext / Google Scholar