Trotz des dramatischen Rückgangs der Morbidität und Mortalität im Zusammenhang mit dem humanen Immundefizienzvirus Typ 1 (HIV-1) nach der Entdeckung der Proteaseinhibitoren und dem Aufkommen der hochaktiven antiretroviralen (ARV) Kombinationstherapie Mitte der 1990er Jahre versagten viele Patienten aufgrund von Resistenzen und / oder Unverträglichkeiten (1). Es war klar, dass mehr ARVs benötigt wurden, die auf verschiedene Schritte im Viruslebenszyklus einwirken, gegen resistente Viren wirksam sind und besser vertragen werden. Die Demonstration der Schlüsselrolle der Chemokinrezeptoren CCR5 und CXCR4 beim HIV-1-Eintritt weckte das Interesse an diesem Prozess als neuem ARV-Ziel (2, 3). CCR5 ist der Co-Rezeptor für die Mehrheit der HIV-1-Stämme, und diese Viren werden als CCR5 tropic (R5) bezeichnet. Virusstämme, die CXCR4 verwenden, werden als CXCR4-tropic (X4) bezeichnet, während Stämme, die beide Rezeptoren verwenden können, dual-tropic (4) sind. Viren von einem Patienten können häufig Mischungen von R5-, X4- und Dual-Tropic-Stämmen enthalten, die zusammen als CXCR4-1 bezeichnet werden.
Die Schlüsselrolle von CCR5 beim HIV-1-Eintritt, gepaart mit dem Nachweis, dass Individuen, die für eine 32-Basenpaar-Deletion im CCR5-Gen (CCR5Δ32) homozygot waren und anschließend kein funktionelles CCR5 exprimieren, in hohem Maße vor einer Infektion mit R5 HIV-1 geschützt waren, konzentrierte sich auf CCR5 als attraktives Ziel (5). Obwohl einige Studien subtile Auswirkungen der CCR5Δ32-Mutation auf die Immunfunktion gezeigt haben, wie z. B. verringerte Entzündungswerte bei Hepatitis C-infizierten Personen und Erholung von Hepatitis B bei Heterozygoten; Während Homozygoten anfälliger für Frühsommer-Meningoenzephalitis und schwere West-Nil-Virus-Krankheit sind, haben diese Personen wenig offensichtliche nachteilige Auswirkungen auf ihre Gesundheit (5, 6). Dies, zusammen mit der Tatsache, dass Mitglieder der G-Protein-gekoppelten Rezeptor-Superfamilie oft auf die Entwicklung potenter, selektiver und oral bioverfügbarer Medikamente zurückzuführen sind (7), führte zur Initiierung von CCR5-Ligandenentdeckungsprogrammen durch mehrere Gruppen, darunter ein Team von Pfizer Global Research and Development mit Sitz in den Sandwich Laboratories im Vereinigten Königreich.
Maraviroc (UK-427.857, MVC) wurde durch Hochdurchsatz-Screening der Pfizer-Wirkstoffbibliothek unter Verwendung eines Chemokin-Radioliganden-Bindungstests entdeckt. Die vielversprechendste Verbindung aus dem Screening-Prozess wurde für die Wirksamkeit gegen den Rezeptor, antivirale Aktivität, pharmakokinetische Eigenschaften und Selektivität gegen menschliche zelluläre Ziele durch eine große medizinische Chemie Anstrengung optimiert, in der fast 1000 Moleküle charakterisiert wurden (7). MVC bindet in der Transmembrantasche von CCR5 und ist ein langsam versetzter funktioneller Antagonist, der die Internalisierung verhindert (7, 8). Es hat eine starke antivirale Aktivität gegen eine Vielzahl von HIV-1-Isolaten (7). Zusammen mit seinem ausgezeichneten präklinischen Sicherheitsprofil und seiner akzeptablen Pharmakokinetik führte dies dazu, dass es im Dezember 2000 als klinischer Kandidat nominiert wurde (7).
Es war immer klar, dass die klinische Entwicklung von CCR5-Antagonisten eine Herausforderung darstellen würde, da dies die ersten auf den Wirt gerichteten ARV-Medikamente sein würden und wir uns daher auf Neuland begeben würden. Um wichtigen Problemen vorzubeugen, wurde sehr bald nach dem Start des Discovery-Programms ein klinisches Entwicklungsteam eingerichtet, und ich wurde als Leiter des frühen Entwicklungsteams eingestellt, das im Februar 1999 zu Pfizer kam. Wir haben mehrere wichtige Herausforderungen identifiziert, die bei der Gestaltung des klinischen Programms zu bewältigen sind, zusätzlich zum Nachweis von Sicherheit und Wirksamkeit. Die erste davon war, dass es keinen kommerziell erhältlichen, klinisch validierten Assay zur Identifizierung von Patienten gab, die mit R5 HIV-1 infiziert waren. Dies war kritisch, da MVC nur gegen R5-HIV-1-Stämme aktiv ist (7). Zweitens blieben trotz des scheinbar gesunden Phänotyps von Personen mit CCR5Δ32 (5, 6) Bedenken hinsichtlich der Sicherheit einer Langzeitexposition gegenüber CCR5-Antagonisten bestehen, da sich die Blockierung von CCR5 von der angeborenen Abwesenheit des CCR5-Rezeptors unterscheiden kann, bei der das Immunsystem in Abwesenheit von CCR5 gereift ist und sich möglicherweise Kompensationsmechanismen entwickelt haben. Schließlich steigt bei HIV-1-infizierten Personen die Inzidenz von CXCR4-verwendenden HIV-1-Stämmen mit fortschreitender Krankheit und Abnahme der CD4-Zellzahl (9), obwohl kein kausaler Zusammenhang zwischen CXCR4-verwendendem Virus und CD4-Zelldepletion nachgewiesen wurde. Dies hat zu Bedenken geführt, dass der selektive Druck eines CCR5-Antagonisten die Viruspopulation dazu bringen könnte, CXCR4 zu verwenden, und zu einem Rückgang der CD4-Zellen führen könnte.
Phase-1-Einzel- und Mehrfachdosisstudien an gesunden Probanden, die 2001 und in der ersten Hälfte des Jahres 2002 durchgeführt wurden, zeigten, dass MVC in Mehrfachdosen von bis zu 300 mg zweimal täglich (BID) sicher und gut verträglich war, ein pharmakokinetisches Profil aufwies, das mit der oralen Dosierung einmal täglich (QD) oder zweimal täglich (BID) kompatibel war, mit anderen ARVs kombiniert werden konnte und dass Dosen von ≥100 mg BID in vitro zu einer Exposition über dem geometrischen Mittel von IC90 führten (7, 10). Zum Nachweis der Pharmakologie wurde die CCR5-Rezeptorsättigung mit einem maßgeschneiderten ex vivo MIP-1β-Internalisierungstest gemessen. Es wurde eine dosisabhängige Sättigung nachgewiesen, wobei Dosen von ≥25 mg QD zu nahezu maximalen Sättigungswerten führten, was die interessante Möglichkeit aufwirft, dass MVC in Dosen von nur 25 mg QD wirksam sein könnte. Die Rezeptorsättigung blieb nach Absetzen der Dosierung mehrere Tage lang hoch, was einen langsamen Offset vom Rezeptor in vivo widerspiegelt (11).
Wir waren beide begeistert und ermutigt von den Phase-1-Daten und gingen schnell zu einem Phase-2a-Proof-of-Concept-Programm über. HIV-1-infizierte Patienten wurden unter Verwendung eines neuartigen phänotypischen Tropismus-Assays (Trofile®, Monogram Biosciences, South San Francisco, CA, USA) nur auf das Vorhandensein des R5-Virus untersucht und erhielten MVC als Monotherapie für 10 Tage (12). Die CCR5-Rezeptorsättigung wurde in dieser Studie gemessen, um die Möglichkeit der Verwendung als Biomarker für die Wirksamkeit und in der therapeutischen Überwachung zu bewerten. Die mit Spannung erwarteten Daten entsprachen unseren Erwartungen und zeigten, dass Dosen von ≥100 mg ZWEIMAL täglich zu einer mittleren maximalen HIV-1-RNA-Reduktion von > 1 führten.5log10 (Abbildung 1A), wobei alle Patienten mit Ausnahme eines Patienten mit X4-Virus, der fälschlicherweise eingeschlossen wurde, eine HIV-1-RNA-Reduktion von mindestens 1log10 erreichten (12). Dies gab uns die Gewissheit, dass der Assay Patienten, die wahrscheinlich auf MVC ansprechen, korrekt identifizierte. Der HIV-1-RNA-Nadir trat 1-5 Tage nach der letzten MVC-Dosis auf, was mit einer verlängerten Rezeptorsättigung übereinstimmte, wie in den Phase-1-Studien gezeigt (12). Für alle Dosen außer 25 mg wurde während des gesamten Dosierungszeitraums eine QD-Rezeptorsättigung von > 80% beobachtet. Es gab jedoch keine Korrelation zwischen der Verringerung der Viruslast und dem Grad der Rezeptorsättigung. Die wahrscheinlichste Erklärung dafür ist, dass für die antivirale Wirksamkeit eine sehr hohe Rezeptorsättigung erforderlich ist und die inhärente Variabilität des Assays keine Differenzierung in diesem Ausmaß zulässt (11, 12).
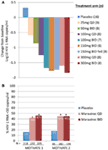
Abbildung 1. Maraviroc Proof of Concept und Phase 3 Wirksamkeitsergebnisse. (A) Mittlere maximale Veränderung der HIV-1-RNA gegenüber dem Ausgangswert bei Patienten, die eine MVC-Monotherapie erhalten. Basierend auf Phase-1-Daten und Modellierung und Simulation wurden Dosen von 25 mg QD bis 300 mg BID (einschließlich 150 mg BID gefüttert und gefastet) ausgewählt. HIV-1-RNA, Sicherheit und MVC-Pharmakokinetik wurden bewertet (12). (B) PHASE 1 und 2 – Anteil der Patienten, die in Woche 48 eine HIV-1-RNA < 50 Kopien / ml erreichten. HIV-1-infizierte Patienten mit R5 HIV-1 und Triple-Class-Erfahrung und / oder Resistenz wurden randomisiert, um MVC QD oder BID oder Placebo in Kombination mit einem optimierten antiretroviralen Hintergrundschema (OBT) zu erhalten. P < 0.001 (13, 14).
Die Phase-2a-Daten sorgten im gesamten Unternehmen für Aufregung, und wir wollten das klinische Entwicklungsprogramm so schnell wie möglich vorantreiben, da ein hoher medizinischer Bedarf an neuen ARVs zur Behandlung von Patienten ohne oder mit eingeschränkten Behandlungsmöglichkeiten bestand. Das umfangreiche Phase-1-Programm (einschließlich mehrerer Arzneimittelwechselwirkungsstudien) und der breite Dosisbereich, die in den Proof-of-Concept-Studien der Phase 2a bewertet wurden, sowie Modellierung und Simulation gaben uns ein sehr gutes Verständnis der wahrscheinlich wirksamen Dosis von MVC in Kombination mit anderen ARVs. Wir konnten daher direkt zu Phase-3-Wirksamkeitsstudien übergehen, in denen MVC bei 300 mg (oder gleichwertig, abhängig von gleichzeitig verabreichten Arzneimitteln) QD und BID untersucht wurde, ohne dass eigenständige Phase-2b-Dosisstudien durchgeführt werden mussten, wodurch der Entwicklungszeitrahmen erheblich verkürzt wurde. Ende 2004 initiierten wir vier große Studien; STUDIE 1 und 2 bei behandlungserfahrenen Patienten mit R5 HIV-1 (13, 14), MERIT (eine Phase-3-Studie mit einem Phase-2b-Roll-in) bei behandlungsnaïven Patienten mit R5 HIV-1 (15) und Studie A4001029, eine Phase-2b-Sicherheitsstudie bei behandlungserfahrenen Patienten mit Nicht-CCR5-Tropenvirus (CXCR4-verwendendes oder nicht phänotypisierbares Virus) (16).
Dies war ein gewaltiges Unterfangen, bei dem 4794 Patienten an mehr als 200 Standorten in den USA, Kanada, Europa, Australien, Südafrika, Mexiko und Argentinien untersucht wurden. Zwei weitere niedermolekulare CCR5-Antagonisten (Aplaviroc und Vicriviroc) wurden zu diesem Zeitpunkt ebenfalls in Phase-2b-Studien untersucht (17, 18). Zusätzlich zu den üblichen Herausforderungen bei der Verwaltung großer klinischer Studien wurden uns zwei Curveballs geworfen, von denen die erste das Absetzen von Aplaviroc aufgrund idiosynkratischer Hepatotoxizität war. Es gab Spekulationen, dass dies ein Klasseneffekt von CCR5-Antagonisten sein könnte, da CCR5-Knockout-Mäuse anfälliger für eine durch Canavalin-A vermittelte Hepatoxizität sind (17). Darüber hinaus entwickelte ein Patient in der MERIT-Studie eine schwere Hepatotoxizität. Die Daten deuteten darauf hin, dass es wahrscheinlich mit Isoniazid oder Cotrimoxazol zusammenhängt, aber eine beitragende Rolle für MVC konnte nicht ausgeschlossen werden (15). Eine eingehende Überprüfung aller Daten auf Hinweise auf Hepatotoxizität bei MVC und ein hohes Maß an Wachsamkeit bei Signalen ergab keine Hinweise auf einen systematischen Anstieg der Leberenzyme oder anderer Marker für Hepatotoxizität. Kurz darauf wurden Bedenken hinsichtlich eines potenziell erhöhten Risikos für bestimmte maligne Erkrankungen nach dem Auftreten von Lymphomen bei vier Patienten, die Vicriviroc in der Studie ACTG5211 erhielten, geäußert (18). Anfänglich gab es Bedenken, dass dies ein Klasseneffekt sein könnte, der auf dem immunmodulatorischen Potenzial von CCR5-Antagonisten beruht, aber die Überprüfung der Daten aus anderen Vicriviroc-Studien sowie der laufenden MVC-Studien stützte diese Theorie nicht (18).
Die Daten aus den STUDIEN 1 und 2 sowie aus A4001029 lagen früher als in MERIT vor, da die Studiendauer bei Studien mit behandlungserfahrenen Patienten in der Regel kürzer ist. Mit großer Spannung warteten wir auf die Zwischenanalysen der MOTIVATE-Studien in Woche 24 im Oktober 2006 und waren begeistert zu sehen, dass signifikant mehr Patienten, die MVC erhielten, eine HIV-1-RNA von < 50 Kopien / ml (der Schlüsselmarker für die Wirksamkeit) aufwiesen als diejenigen, die Placebo OBT erhielten. Dies wurde durch die Daten aus Woche 48 bestätigt, die die Dauerhaftigkeit des Ansprechens belegen (Abbildung 1B) (13, 14). Im Gegensatz dazu schienen Patienten mit nicht-CCR5-tropischem HIV-1, die MVC in A4001029 erhielten, im Vergleich zu Placebo keinen signifikanten virologischen Nutzen zu erzielen (16). Die Analyse der Sicherheitsdaten ergab keine wesentlichen Bedenken. Insbesondere gab es keine Hinweise auf eine nachteilige Wirkung auf die Immunfunktion, ohne Zunahme von Infektionsepisoden oder Malignomen bei mit MVC behandelten Patienten. Die Beurteilung des Virustropismus beim Versagen zeigte, dass > 50% der Patienten, bei denen die MVC-Therapie versagte, bei Versagen ein CXCR4-verwendendes Virus aufwiesen, es gab jedoch keine Hinweise auf eine schädliche Wirkung auf die CD4-Zellzahl (14). Die virologische Beurteilung zeigte, dass das CXCR4-verwendende Virus, das unter selektivem MVC-Druck auftrat, aus einer bereits bestehenden Minderheitenpopulation stammte und nicht de novo auftrat (19). Insgesamt zeigten diese Ergebnisse deutlich den Nutzen von MVC bei der Behandlung von behandlungserfahrenen Patienten mit R5 HIV-1. Eine große Anstrengung des Teams führte zur Einreichung von Dossiers zur Registrierung sowohl in den USA als auch in Europa, nur 2 Monate nachdem die Zwischendaten verfügbar wurden. MVC (300 mg BID) erhielt die Zulassung für die Verwendung (in Kombination mit anderen ARVs) in den USA im August 2007 nur 6.5 jahre nachdem es als Kandidat für die klinische Entwicklung nominiert wurde. Einen Monat später wurde es auch für die Anwendung in dieser Population in der EU zugelassen.
Die Analyse der MERIT-Studie in Woche 48 war enttäuschend, da MVC plus Zidovudin/Lamivudin (HIV-1-RNA <50 Kopien/ml, 65,3%) die voreingestellten Kriterien für die Nichtunterlegenheit (untere Grenze des 1-seitigen 97,5-Konfidenzintervalls unter -10%) gegenüber Efavirenz plus Zidovudin/Lamivudin (HIV-1-RNA <50 Kopien/ml, 65,3 mL, 69,3%) (15). Die Patienten für diese Studie wurden jedoch mit dem ursprünglichen Trofile-Assay auf R5-Virus untersucht. Dieser Test wurde in der Zwischenzeit verbessert, um empfindlicher für den Nachweis von Minderheitenpopulationen des CXCR4-verwendenden Virus zu sein. Alle Screening-Proben für Patienten in MERIT wurden anschließend mit dem erweiterten Assay erneut getestet und eine Post-hoc-Analyse durchgeführt, bei der nur Patienten mit R5-Virus durch den empfindlicheren Assay eingeschlossen wurden. In dieser Analyse betrugen die Ansprechraten für MVC und Efavirenz 68,3 bzw. 68,5%, wobei die untere Grenze des 97,5%-Konfidenzintervalls über -10% lag (15). Basierend auf diesen Daten wurde MVC im November 2009 von der US-amerikanischen Food and Drug Administration auch für die Anwendung bei therapienaïven Patienten zugelassen.
MVC hat sich nicht nur als wertvolle Ergänzung des ständig wachsenden ARV-Arzneimittel-Armamentariums erwiesen, sondern die Daten aus diesen Studien haben unser Verständnis des HIV-Tropismus und der Beziehung zwischen Tropismus und Krankheitsprogression verbessert. Für mich persönlich war dies eine Zeit großer Aufregung und Zufriedenheit, sowohl als Arzt als auch als Wissenschaftler.
Autorenbeiträge
ER entwarf dieses Manuskript basierend auf ihren persönlichen Erfahrungen als Mitglied des MVC-Entwicklungsteams. Alle Daten wurden an anderer Stelle vollständig veröffentlicht. Alle Studien wurden in Übereinstimmung mit den Grundsätzen der Erklärung von Helsinki und mit allen Internationalen Konferenz über die Harmonisierung Gute klinische Praxis-Richtlinien und lokalen regulatorischen und rechtlichen Anforderungen durchgeführt. Alle Studien wurden von unabhängigen Ethikkommissionen genehmigt und alle Patienten gaben eine schriftliche Einverständniserklärung ab.
Erklärung zu Interessenkonflikten
Elna Van Der Ryst war zum Zeitpunkt der Entwicklung von MVC Mitarbeiterin von Pfizer Global Research and Development. Derzeit berät sie Pfizer.
Danksagung
Ich möchte den Kollegen von Pfizer danken, die zum MVC-Entdeckungs- und Entwicklungsprogramm beigetragen haben, sowie den Forschern und Patienten, die an den klinischen Studien teilgenommen haben.
1. Richman DD, Morton SC, Wrin T, Hellmann N, Berry S, Schapiro MF, et al. Die Prävalenz der antiretroviralen Arzneimittelresistenz in den Vereinigten Staaten. AIDS (2004) 18:1393-401. doi: 10.1097/01.AIDS.0000131310.52526.c7
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
2. Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, et al. Der Eintritt von HIV-1 in CD4 + -Zellen wird durch den Chemokinrezeptor CC-CKR-5 vermittelt. Natur (1996) 381: 667-73. doi:10.1038/381667a0
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
3. Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1-Eintrittskofaktor: funktionelle cDNA-Klonierung eines sieben-transmembranen, G-Protein-gekoppelten Rezeptors. Wissenschaft (1996) 272: 872-7. doi:10.1126/Wissenschaft.272.5263.872
CrossRef Volltext / Google Scholar
4. Berger EA, Doms RW, Fenyö EM, Korber BT, Littman DR, Moore JP, et al. Eine neue Klassifikation für HIV-1. Natur (1998) 391: 240. doi:10.1038/34571
CrossRef Volltext / Google Scholar
5. Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber C, et al. Resistenz gegen HIV-1-Infektion bei kaukasischen Individuen, die mutierte Allele des CCR5-Chemokinrezeptorgens tragen. Natur (1996) 382: 722-5. doi:10.1038/382722a0
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
6. Gilliam BL, Riedel DJ, Redfield RR. Klinische Verwendung von CCR5-Inhibitoren bei HIV und darüber hinaus. J Transl Med (2011) 9(Ergänzung 1):S9. doi:10.1186/1479-5876-9-S1-S9
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
7. Dorr P., Westby M., Dobbs S., Griffin P., Irvine B., Macartney M., et al. Maraviroc (UK-427.857), ein potenter, oral bioverfügbarer und selektiver niedermolekularer Inhibitor des Chemokinrezeptors CCR5 mit Breitspektrum-Anti-Human-Immundefizienz-Virus-Typ-1-Aktivität. Antimikrobielle Mittel Chemother (2005) 49: 4721-32. doi:10.1128/AAC.49.11.4721-4732.2005
CrossRef Volltext / Google Scholar
8. Kondru R, Zhang J, Ji C, Mirzadegan T, Rotstein D, Sankuratri S, et al. Molekulare Wechselwirkungen von CCR5 mit Hauptklassen von niedermolekularen Anti-HIV-1-CCR5-Antagonisten. Mol Pharmacol (2008) 73:789-800. doi: 10,1124/mol.107.042101
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
9. Berger EA, Murphy PM, Farber JM. Chemokinrezeptoren als HIV-1-Co-Rezeptoren: Rollen beim Viruseintritt, Tropismus und Krankheit. Annu Rev Immunol (1999) 17:657-700. doi:10.1146/Jahrbuch.immunol.17.1.657
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
10. Abel S., van der Ryst E., Rosario MC, James I., Ridgway C., Medhurst C., et al. Bewertung der Pharmakokinetik, Sicherheit und Verträglichkeit von Maraviroc, einem neuartigen CCR5-Antagonisten, bei gesunden Probanden. Br J Clin Pharmacol (2008) 65(S1):5-18. Ursprungsbezeichnung:10.1111/j.1365-2125.2008.03130.x
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
11. Rosario MC, Jacqmin P, Dorr P, James I, Jenkins T, Abel S, et al. Pharmakokinetisch-pharmakodynamische Analyse der CCR5-Rezeptorbelegung durch Maraviroc bei gesunden Probanden und HIV-positiven Patienten. Br J Clin Pharmacol (2008) 65(S1):86-94. Ursprungsbezeichnung:10.1111/j.1365-2125.2008.03140.x
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
12. Fätkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AIM, et al. Wirksamkeit der Kurzzeitmonotherapie mit Maraviroc, einem neuen CCR5-Antagonisten bei HIV-1-infizierten Patienten. Nat Med (2005) 11:1170-2. doi:10.1038/nm1319
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
13. Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, et al. Maraviroc für zuvor behandelte Patienten mit R5 HIV-1-Infektion. N Engl J Med (2008) 359:1429-41. doi:10.1056/NEJMoa0803152
CrossRef Volltext / Google Scholar
14. Fätkenheuer G, Nelson M, Lazzarin A, Konourina I, Hoepelman IM, Lampiris H, et al. Subgruppenanalysen von Maraviroc bei vorbehandelter R5-HIV-1-Infektion. N Engl J Med (2008) 359:1442-55. doi:10.1056/NEJMoa0803154
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
15. Cooper DA, Heera J, Goodrich J, Tawadrous M, Saag M, DeJesus E, et al. Maraviroc versus Efavirenz, beide in Kombination mit Zidovudin/Lamivudin, zur Behandlung von antiretroviral-naïven Patienten mit CCR5-tropischem HIV-1. J Infizieren Dis (2010) 201: 803-13. doi:10.1086/650697
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
16. Saag M, Goodrich J, Fätkenheuer G, Clotet B, Clumeck N, Sullivan J, et al. Eine doppelblinde, placebokontrollierte Studie mit Maraviroc bei behandlungserfahrenen Patienten, die mit nicht-CCR5-tropischem HIV-1 infiziert waren: 24-Wochen-Ergebnisse. J Infizieren Dis (2009) 11: 1638-47. doi:10.1086/598965
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
17. Nichols WG, Stahl HM, Bonny T, Adkison K, Curtis L, Millard J, et al. Hepatotoxizität beobachtet in klinischen Studien mit Aplaviroc (GW873140). Antimikrobielle Mittel Chemother (2008) 52: 858-65. doi:10.1128/AAC.00821-07
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
18. Tsibris AM, Paredes R, Chadburn A, Su Z, Henrich TJ, Krambrink A, et al. Lymphomdiagnose und Plasma-Epstein-Barr-Viruslast während der Vicriviroc-Therapie: Ergebnisse der AIDS Clinical Trials Group A5211. Clin Infizieren Dis (2009) 48: 642-9. doi:10.1086/597007
Pubmed Zusammenfassung / Pubmed Volltext / CrossRef Volltext / Google Scholar
19. Es sind keine frei zugänglichen ergänzenden Informationen verfügbar in: Lewis M, Simpson P, Fransen S, Huang W, Whitcomb J, Mosley M, et al. Das in den Phase-III-Studien MOTIVATE 1 und 2 bei Patienten, die Maraviroc erhielten, nachgewiesene CXCR4-nutzende Virus stammt von einer bereits bestehenden Minderheit von CXCR4-nutzenden Viren. Antivir Ther (2007) 12:S65.
Google Scholar