- Introduction
- Methoden
- Filterung von ribosomalen Profilierungsdaten
- Virtuelle Genome zusammensetzen
- Bestimmung der Ribo-seq Read-Mapped Region an einer Kreuzung (RMRJ) von circRNAs
- Training des Modells und Klassifizierung von RMRJs
- Vorhersage von translatierten Peptiden durch RMRJs
- Ergebnisse und Diskussion
- Translatierte circRNAs bei Menschen und A. thaliana
- Funktionelle Anreicherung humaner und A. thaliana circRNAs mit Kodierungspotential
- Genauigkeitstest für CircCode
- Einfluss der Sequenziertiefe der Ribo-seq-Daten
- Vergleich von CircCode mit anderen Tools
- Schlussfolgerungen
- Verfügbarkeit und Anforderungen
- Datenverfügbarkeitserklärung
- Autorenbeiträge
- Finanzierung
- Interessenkonflikt
- Ergänzungsmaterial
Introduction
Zirkuläre RNAs (circRNAs) sind eine spezielle Art von nichtkodierendem RNA-Molekül, das zu einem heißen Forschungsthema auf dem Gebiet der RNA geworden ist und viel Aufmerksamkeit erhält (Chen und Yang, 2015). Im Vergleich zu herkömmlichen linearen RNAs (mit 5′- und 3′-Enden) haben circRNA-Moleküle normalerweise eine geschlossene kreisförmige Struktur; Dies macht sie stabiler und weniger anfällig für Abbau (Vicens und Westhof, 2014). Obwohl die Existenz von circRNAs seit einiger Zeit bekannt ist, wurden diese Moleküle als Nebenprodukt des RNA-Spleißens angesehen. Mit der Entwicklung von Hochdurchsatzsequenzierungs- und Bioinformatiktechnologien sind circRNAs jedoch in Tieren und Pflanzen weithin anerkannt (Chen und Yang, 2015). Neuere Studien haben auch gezeigt, dass eine große Anzahl von circRNAs in Zellen in kleine Peptide übersetzt werden kann (Pamudurti et al., 2017) und spielen trotz ihrer manchmal geringen Expression eine Schlüsselrolle (Hsu und Benfey, 2018; Yang et al., 2018). Obwohl immer mehr circRNAs identifiziert werden, müssen ihre Funktionen in Pflanzen und Tieren im Allgemeinen noch untersucht werden. Zusätzlich zu ihren Funktionen als miRNA-Lockvögel haben circRNAs ein wichtiges Translationspotenzial, aber es stehen keine Werkzeuge zur Verfügung, um die Translationsfähigkeiten dieser Moleküle spezifisch vorherzusagen (Jakobi und Dieterich, 2019).
Es existieren mehrere Werkzeuge zur Vorhersage und Identifizierung von circRNAs, wie CIRI (Gao et al., 2015), CIRCexplorer (Dong et al., 2019), CircPro (Meng et al., 2017) und circtools (Jakobi et al., 2018). Unter ihnen kann CircPro übersetzte circRNAs aufdecken, indem es einen Translationspotential-Score für circRNAs basierend auf CPC berechnet (Kong et al., 2007), die ein Werkzeug zur Identifizierung des offenen Leserahmens (ORF) in einer bestimmten Reihenfolge ist. Da jedoch einige circRNAs das Startcodon während der Translation nicht verwenden (Ingolia et al., 2011; Slavoff et al., 2013; Kearse und Wilusz, 2017; Spealman et al., 2018), kann der Einsatz von CPC einige wirklich übersetzte circRNAs herausfiltern. In dieser Studie verwendeten wir BASiNET (Ito et al., 2018), einem RNA-Klassifikator, der auf den Methoden des maschinellen Lernens (Random Forest und J48-Modell) basiert. Es transformiert zunächst die gegebenen codierenden RNAs (positive Daten) und nicht-codierenden RNAs (negative Daten) und stellt sie als komplexe Netzwerke dar; Es extrahiert dann die topologischen Maße dieser Netzwerke und konstruiert einen Merkmalsvektor, um das Modell zu trainieren, das zur Klassifizierung der Codierungskapazität von circRNAs verwendet wird. Mit dieser Methode wird eine fehlerhafte Filterung von translatierten circRNAs, die nicht durch AUG initiiert werden, vermieden. Zusätzlich wurde die Ribo-seq-Technologie, die auf Hochdurchsatzsequenzierung zur Überwachung von rPFS (Ribosomal protected Fragments) von Transkripten basiert (Guttman et al., 2013; Brar und Weissman, 2015) können verwendet werden, um die Positionen von circRNAs zu bestimmen, die übersetzt werden (Michel und Baranov, 2013). Um die Codierungsfähigkeit von circRNAs zu identifizieren, haben wir das Tool CircCode entwickelt, das ein Python 3–basiertes Framework beinhaltet, und CircCode angewendet, um das Translationspotenzial von circRNAs von Menschen und Arabidopsis thaliana zu untersuchen. Unsere Arbeit bietet eine reichhaltige Ressource für die weitere Untersuchung der Funktionen von circRNAs mit Codierungskapazität.
Methoden
CircCode wurde in der Programmiersprache Python 3 geschrieben; es verwendet Trimmomatic (Bolger et al., 2014), bowtie (Langmead und Salzberg, 2012) und STAR (Dobin et al., 2013), um rohe Ribo-Seq-Lesevorgänge zu filtern und diese gefilterten Lesevorgänge dem Genom zuzuordnen. CircCode identifiziert dann Ribo-seq-Read-Mapped-Regionen in circRNAs, die Junctions enthalten. Danach werden die in den circRNAs abgebildeten Kandidaten-Sequenzen nach Klassifikatoren (J48-Modell) in codierende RNAs und nicht-codierende RNAs nach BASiNET sortiert. Schließlich werden kurze Peptide, die durch Translation erzeugt werden, als potentielle kodierende Regionen von circRNAs identifiziert. Der gesamte Prozess von CircCode besteht aus fünf Schritten (Abbildung 1).
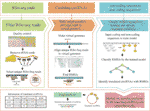
Abbildung 1 Der Workflow von CircCode. Die oberste Ebene stellt die Eingabedatei dar, die für jeden Schritt von CircCode erforderlich ist. Die mittlere Schicht ist in drei Teile unterteilt, und jeder Teil stellt eine andere Betriebsstufe dar. Von links nach rechts stellt der erste Teil die Filterung der Ribo-seq-Daten dar; Die Qualitätskontrolle wird von Trimmomatic ausgeführt, und die rRNA-Lesevorgänge werden von Bowtie entfernt. Der zweite Teil stellt die Schritte dar, die verwendet werden, um das virtuelle Genom zu erzeugen und die gefilterten Lesevorgänge mit STAR auf das virtuelle Genom auszurichten. Der letzte Teil stellt die Identifizierung von übersetzten circRNAs durch maschinelles Lernen dar. Die untere Schicht stellt den letzten Schritt dar, der zur Vorhersage der aus den circRNAs übersetzten Peptide und der endgültigen Ausgabeergebnisse verwendet wird, einschließlich Informationen zu übersetzten circRNAs und ihren Translationsprodukten.
Filterung von ribosomalen Profilierungsdaten
Zunächst werden Fragmente und Adapter von geringer Qualität in den Ribo-Seq-Lesevorgängen von Trimmomatic mit den Standardparametern entfernt, um saubere Ribo-Seq-Lesevorgänge zu erhalten. Zweitens werden diese sauberen Ribo-Seq-Lesevorgänge einer rRNA-Bibliothek zugeordnet, um von rRNA abgeleitete Lesevorgänge mithilfe von Bowtie zu entfernen. Da die Leselängen von Ribo-seq relativ kurz sind (im Allgemeinen weniger als 50 bp), ist es möglich, dass ein Lesevorgang mehreren Regionen entspricht. In diesem Fall ist es schwierig zu bestimmen, welcher Region ein bestimmter Lesevorgang entspricht. Um dies zu vermeiden, werden die sauberen Ribo-Seq-Lesevorgänge auf das Genom einer interessierenden Spezies abgebildet, und die Lesevorgänge, die nicht perfekt auf das Genom ausgerichtet sind, werden als die endgültigen eindeutigen Ribo-Seq-Lesevorgänge angesehen.
Virtuelle Genome zusammensetzen
CircRNAs erscheinen normalerweise als ringförmige Moleküle in Eukaryoten und können anhand ihrer Rückspleißverbindungen identifiziert werden. Die Sequenzen von circRNAs in der Fasta-Datei liegen jedoch häufig in linearer Form vor. Theoretisch zeigt das Ergebnis an, dass der Übergang zwischen dem 5′-terminalen Nukleotid und dem 3′-terminalen Nukleotid liegt, obwohl der Übergang und die Sequenz in der Nähe des Übergangs nicht direkt betrachtet werden können, wodurch Ribo-seq-Lesevorgänge auf einfache Weise an circRNA-Sequenzen, einschließlich Übergängen, ausgerichtet werden.
CircCode verbindet die Sequenz jeder circRNA im Tandem, so dass die Kreuzung für jede in der Mitte der neu konstruierten Sequenz liegt. Wir trennten auch jede Serieneinheit durch 100 N Nukleotide, um Verwechslungen beim Sequenzausrichtungsschritt zu vermeiden (die Länge jedes RPF beträgt weniger als 50 bp). Schließlich erhielten wir ein virtuelles Genom, das nur aus Kandidaten-circRNAs im Tandem bestand, die durch 100 Ns getrennt waren. Da sich CircCode nur auf die Ausrichtung zwischen Ribo-Seq-Lesevorgängen und circRNA-Sequenzen konzentriert, können wir das Codierungspotential von circRNAs untersuchen, indem wir die Ribo-Seq-Lesevorgänge auf dieses virtuelle Genom abbilden, was eine große Menge an Rechenzeit einsparen kann (das virtuelle Genom ist viel kleiner als das gesamte Genom) und Erhöhen Sie die Genauigkeit (durch Vermeidung von Interferenzen zwischen Upstream- und Downstream-Sequenzvergleichen der circRNAs).
Bestimmung der Ribo-seq Read-Mapped Region an einer Kreuzung (RMRJ) von circRNAs
Die endgültigen eindeutigen Ribo-seq-Reads werden mit STAR auf ein zuvor erstelltes virtuelles Genom abgebildet. Da jede Tandem-circRNA-Einheit vor der Herstellung des virtuellen Genoms durch 100 N-Basen getrennt wurde, wurde die größte Intronlänge mit dem Parameter „–alignIntronMax 10″ auf nicht mehr als 10 Basen eingestellt.“ Dieser Parameter eliminiert jegliche Interaktion zwischen verschiedenen circRNAs in der Sequenzausrichtung. Im zweiten Schritt der virtuellen Genomproduktion speichert CircCode Positionsübergangsinformationen für jede circRNA im virtuellen Genom. Wenn die Ribo-Seq-Lese-kartierte Region im virtuellen Genom die Kreuzung der circRNA umfasst und die Anzahl der kartierten Ribo-Seq-Lesevorgänge an der Kreuzung (NMJ) größer als 3 ist, kann die Ribo-Seq-Lese-kartierte Region an der Kreuzung der circRNAs als RMRJ angesehen werden, die ein grob übersetztes Segment von circRNAs in der Nähe der Verbindungsstelle aufdeckt.
Training des Modells und Klassifizierung von RMRJs
Obwohl RMRJs einen starken Übersetzungsnachweis darstellen können, gibt es bei dieser Methode immer noch einige Mängel. Da die Länge der Lesevorgänge der ribosomalen Karte kurz ist, kann ein Lesevorgang mit der falschen Position verglichen werden. Daher ist es nicht überzeugend, die von den Ribo-seq-Lesungen abgedeckte Region einfach als übersetzte Region zu betrachten. Zu diesem Zweck wird die Methode des maschinellen Lernens verwendet, um die Codierungsfähigkeit des RMRJ zu identifizieren. Zunächst extrahiert CircCode kodierende RNAs (positive Daten) und nicht-kodierende RNAs (negative Daten) aus einer Spezies von Interesse und verwendet sie für das Modelltraining anhand der Differenz der Merkmalsvektoren zwischen kodierenden und nicht-kodierenden RNAs. CircCode verwendet dann das trainierte Modell, um die im vorherigen Schritt von BASiNET erhaltenen RMRJs zu klassifizieren. Wird die RMRJ einer circRNA als kodierende RNA erkannt, so kann diese circRNA als translatierte circRNA identifiziert werden.
Vorhersage von translatierten Peptiden durch RMRJs
Da die Expression von circRNAs in Organismen gering ist, zeigen Ribo-seq-Daten die genaue 3-nt-Periodizität bei weniger rPFS nicht eindeutig. Daher ist es schwierig, die genaue Übersetzungsstartstelle einer übersetzten circRNA zu bestimmen. Aufgrund des Vorhandenseins eines Stop-Codons in einigen RMRJs und weil das Start-Codon schwer zu bestimmen ist, ist das Verfahren zum Finden eines ORF basierend auf einem Start-Codon und einem Stop-Codon nicht durchführbar.
Um die wahren Translationsregionen dieser circRNAs zu bestimmen und das endgültige Translationsprodukt zu erzeugen, wurde FragGeneScan (Rho et al., 2010), das proteinkodierende Regionen in fragmentierten Genen und Genen mit Frameshifts vorhersagen kann, wird verwendet, um die von circRNAs produzierten translatierten Peptide zu bestimmen.
Um den umständlichen Ablauf zu vermeiden, können alle Modelle von einem Shell-Skript aufgerufen werden; Der Benutzer kann einfach die angegebene Konfigurationsdatei ausfüllen und in das Skript eingeben, und der gesamte Prozess zur Vorhersage der übersetzten circRNAs wird dann ausgeführt. Darüber hinaus kann CircCode Schritt für Schritt separat ausgeführt werden, sodass der Benutzer die Parameter in der Mitte des Vorgangs anpassen und die Ergebnisse jedes Schritts wie gewünscht anzeigen kann.
Ergebnisse und Diskussion
Nach dem Testen auf mehreren Computern wurde festgestellt, dass CircCode erfolgreich ausgeführt wird und die erforderlichen Abhängigkeiten installiert sind. Um die Leistung von CircCode zu testen, verwendeten wir Daten für Menschen und A. thaliana, um circRNAs mit Translationspotential vorherzusagen. Die Ergebnisse wurden mit circRNAs verglichen, die experimentell als Bestätigung verifiziert wurden. Danach haben wir den Wert der False Discovery Rate (FDR) von CircCode weiter getestet. Wir verwendeten GenRGenS (Ponty et al., 2006), um einen Datensatz für Tests basierend auf bekannten translatierten circRNAs zu generieren und zu bestätigen, dass der FDR-Wert in einem akzeptablen Bereich und auf einem niedrigen Niveau lag. Schließlich bewerteten wir die Auswirkungen verschiedener Sequenzierungstiefen von Ribo-seq-Daten auf CircCode-Vorhersagen und verglichen CircCode mit anderer Software.
Translatierte circRNAs bei Menschen und A. thaliana
Um das CircCode-Tool auf reale Daten anzuwenden, haben wir zuerst die Dateien einschließlich des menschlichen Referenzgenoms GRCh38, der Genomanmerkung und der menschlichen rRNA von Ensembl heruntergeladen. Für A. thaliana wurden die Referenzgenome (TAIR10), Genomanmerkungsdateien und entsprechende rRNA-Sequenzen alle von Ensembl-Pflanzen heruntergeladen. Die Ribo-seq-Daten für Menschen und A. thaliana wurden von RPFdb heruntergeladen (Zugangsnummern: GSE96643, GSE81295, GSE88794) (Hsu et al., 2016; Willems et al., 2017), und alle Kandidaten circRNAs von Mensch und A. thaliana wurden von CIRCPedia v2 (Dong et al., 2018) und PlantcircBase (Chu et al., 2017). Letztendlich identifizierten wir 3.610 translatierte circRNAs vom Menschen und 1.569 translatierte circRNAs von A. thaliana mit CircCode (Ergänzende Daten 1).
Funktionelle Anreicherung humaner und A. thaliana circRNAs mit Kodierungspotential
Unter Verwendung der CircCode-Ergebnisse für human und A. thaliana, dem Online-Tool KOBAS 3.0 (Wu et al., 2006) wurde verwendet, um diese übersetzten circRNAs basierend auf ihren Elterngenen zu kommentieren. Darüber hinaus führten wir eine GO-Funktionsanalyse (Gene Ontology) und eine KEGG-Anreicherungsanalyse (Kyoto Encyclopedia of Genes and Genomes) für diese translatierten circRNAs mit dem R-Paket clusterProfiler (Yu et al., 2012).
Die KEGG-Ergebnisse zeigten, dass die humanen circRNAs bei der Proteinverarbeitung im endoplasmatischen Retikulum-, Kohlenstoffmetabolismus- und RNA-Transportweg angereichert waren. Die GO-Analyse zeigte die Beteiligung humaner translatierter circRNAs an der Regulation der Molekülbindung, der ATPase-Aktivität und anderer biologischer Prozesse im Zusammenhang mit dem RNA-Spleißen. Darüber hinaus sind die translatierten circRNAs von A. thaliana in Signalwegen angereichert, die mit der Stressresistenz zusammenhängen, was darauf hindeutet, dass sie in diesem Prozess eine wichtige Rolle spielen (ergänzende Daten 2).
Genauigkeitstest für CircCode
Um die Genauigkeit von CircCode zu untersuchen, wurden von GenRGenS generierte Testsequenzen verwendet, die das Hidden-Markov-Modell verwenden, um Sequenzen zu erzeugen, die dieselben Sequenzeigenschaften aufweisen (z. B. die Frequenzen verschiedener Nukleotide, verschiedener Codons und verschiedener Nukleotide zu Beginn der Sequenz).
Für diese Studie verwendeten wir zuvor veröffentlichte humane translatierte circRNAs (Yang et al., 2017) als Eingabe für GenRGenS und generierte 10.000 Sequenzen zum Testen von CircCode. Wir wiederholten den Test 10 Mal, und im Durchschnitt wurden 27 übersetzte circRNAs jedes Mal vorhergesagt. Der FDR-Wert wurde mit 0,0027 berechnet, was viel weniger als 0,05 ist, was darauf hinweist, dass die vorhergesagten Ergebnisse glaubwürdig sind.
Zusätzlich verglichen wir die translatierten circRNAs von Menschen, wie sie durch CircCode identifiziert wurden, mit verifizierten Polysom-assoziierten circRNA-Daten (Yang et al., 2017). Unter ihnen wurden 60% der circRNAs durch CircCode identifiziert (ergänzende Daten 3).
Einfluss der Sequenziertiefe der Ribo-seq-Daten
Um den Einfluss der Sequenziertiefe der Ribo-seq-Daten auf die CircCode-Identifikationsergebnisse zu untersuchen, haben wir zunächst den Effekt der Sequenziertiefe auf die Anzahl der translatierten circRNAs getestet (Abbildung 2A). Wenn die Sequenzierungstiefe niedrig war, war die vorhergesagte Anzahl der translatierten circRNAs niedrig, und die Anzahl der translatierten circRNAs nahm mit zunehmender Sequenzierungstiefe zu. Die Anzahl der translatierten circRNAs wurde stabil, wenn die Sequenzierungstiefe nicht weniger als 10 × lineare Transkriptabdeckung erreichte.

Abbildung 2 (A) Auswirkung der Ribo-seq-Datensequenzierungstiefe auf die vorhergesagte Anzahl translatierter circRNAs. (B) Die Auswirkung der Junction Read Number (JRN) auf die CircCode-Empfindlichkeit bei verschiedenen Sequenzierungstiefen.
Zweitens wurde der Einfluss von NMJ auf die Empfindlichkeit bei verschiedenen Sequenziertiefen ebenfalls bewertet (Abbildung 2B). Die Ergebnisse zeigten, dass NMJ mit zunehmender Sequenzierungstiefe weniger Einfluss auf die Empfindlichkeit hatte. CircCode hatte auch eine höhere Empfindlichkeit bei Verwendung von Ribo-seq-Daten mit höherer Sequenzierungstiefe.
Vergleich von CircCode mit anderen Tools
Um CircCode mit anderen Tools wie CircPro zu vergleichen, wurde derselbe Satz von Ribo-Seq-Daten (SRR3495999) von A. thaliana verwendet, um übersetzte circRNAs mit sechs Prozessoren mit 16 Gigabyte RAM zu identifizieren. CircPro identifizierte 44 übersetzte circRNAs in 13 min, während CircCode 76 übersetzte circRNAs in 20 min identifizierte. Daher ist CircCode auf derselben Computerhardwareebene empfindlicher als CircPro, benötigt jedoch mehr Zeit. CircPro ist prägnant und weniger zeitaufwendig als CircCode, aber CircCode kann mehr circRNAs mit Codierungsfähigkeit identifizieren als CircPro.
Schlussfolgerungen
CircRNAs spielen eine wichtige Rolle in der Biologie, und es ist entscheidend, circRNAs mit Kodierungsfähigkeit für die nachfolgende Forschung genau zu identifizieren. Basierend auf Python 3 haben wir CircCode entwickelt, ein benutzerfreundliches Befehlszeilentool mit hoher Empfindlichkeit zur Identifizierung übersetzter circRNAs aus Ribo-Seq-Reads mit hoher Genauigkeit. CircCode zeigt sowohl bei Pflanzen als auch bei Tieren eine gute Leistung. Zukünftige Arbeiten werden die nachgelagerte Charakteranalyse zu CircCode hinzufügen, indem jeder Schritt im Prozess visualisiert und die Genauigkeit der Vorhersage optimiert wird.
Verfügbarkeit und Anforderungen
CircCode ist verfügbar unter https://github.com/PSSUN/CircCode; Betriebssystem (e): Linux, Programmiersprachen: Python 3 und R; weitere Anforderungen: bedtools (Version 2.20.0 oder höher), Bowtie, STAR, Python 3-Pakete (Biopython, Pandas, rpy2), R-Pakete (BASiNET, Biostrings). Die Installationspakete für die benötigte Software sind auf der CircCode Homepage verfügbar. Benutzer müssen sie nicht einzeln herunterladen. Die CircCode-Homepage enthält auch detaillierte Benutzerhandbücher als Referenz. Das Tool ist frei verfügbar. Es gibt keine Nutzungsbeschränkungen für Nichtakademiker.
Datenverfügbarkeitserklärung
Alle relevanten Daten befinden sich im Manuskript und seinen unterstützenden Informationsdateien.
Autorenbeiträge
Konzeption: PS, GL. Datenpflege: PS, GL. Formale Analyse: PS, GL. Schrift – Originalentwurf: PS, GL. Schreiben – Rezension und Bearbeitung: PS, GL.
Finanzierung
Diese Arbeit wurde durch Zuschüsse der National Natural Science Foundation of China (Grant Nr. 31770333, 31370329 und 11631012), des Programms für New Century Excellent Talents in University (NCET-12-0896) und der Fundamental Research Funds for the Central Universities (Nr. GK201403004). Die Förderstellen hatten keine Rolle bei der Studie, ihrem Design, der Datenerhebung und -analyse, der Entscheidung zur Veröffentlichung oder der Erstellung des Manuskripts. Die Geldgeber hatten keine Rolle beim Studiendesign, der Datenerhebung und -analyse, der Entscheidung zur Veröffentlichung oder der Vorbereitung des Manuskripts.
Interessenkonflikt
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Ergänzungsmaterial
Das Ergänzungsmaterial zu diesem Artikel finden Sie online unter: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
Ergänzende Daten 1 / Die Sequenz der vorhergesagten translatierten circRNA und des kurzen Peptids.
Ergänzende Daten 2 / GO-Anreicherung und KEGG-Anreicherung Ergebnisse für Menschen und Arabidopsis thaliana.
Ergänzende Daten 3 / Vergleich von vorhergesagten translatierten circRNAs mit validierten translatierten circRNAs.
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: ein flexibler Trimmer für Illumina-Sequenzdaten. Bioinformatik 30, 2114-2120. doi: 10.1093/bioinformatik/btu170
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Brar, G. A., Weissman, J. S. (2015). Ribosomenprofilierung zeigt das Was, wann, wo und wie der Proteinsynthese. Nat. In: Rev. Mol. In: Cell Biol. 16, 651–664. doi: 10.1038/nrm4069
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Chen, L.-L., Yang, L. (2015). Regulation der circRNA-Biogenese. In: RNA Biol. 12, 381–388. doi: 10.1080/15476286.2015.1020271
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Chu, Q., Zhang, X., Zhu, X., Liu, C., Mao, L., Ye, C., et al. (2017). PlantcircBase: eine Datenbank für pflanzliche zirkuläre RNAs. Mol. Pflanze 10, 1126-1128. Ursprungsbezeichnung: 10.1016/j.molp.2017.03.003
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultraschneller universeller RNA-seq Aligner. Bioinformatik 29, 15-21. doi: 10.1093/bioinformatik/bts635
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Dong, R., Ma, X.-K., Chen, L.-L., Yang, L. (2019). „Genomweite Annotation von circRNAs und deren alternatives Rückspleißen / Spleißen mit der CIRCexplorer-Pipeline“, in Epitranscriptomics. Eds. Wajapeyee, N., Gupta, R. (New York, NY: Springer New York), 137-149. doi: 10.1007/978-1-4939-8808-2_10
CrossRef Volltext / Google Scholar
Dong, R., Ma, X.-K., Li, G.-W., Yang, L. (2018). CIRCpedia v2: eine aktualisierte Datenbank für umfassende zirkuläre RNA-Annotation und Expressionsvergleich. In: Genomics Proteomics Bioinf. 16, 226–233. doi: 10.1016/j.gpb.2018.08.001
CrossRef Volltext / Google Scholar
Gao, Y., Wang, J., Zhao, F. (2015). CIRI: ein effizienter und unvoreingenommener Algorithmus zur De-Novo-zirkulären RNA-Identifizierung. In: Genome Biol. 16, 4. ust-IDNR.: 10.1186/s13059-014-0571-3
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Guttman, M., Russell, P., Ingolia, NT, Weissman, JS, Lander, ES (2013). Die Ribosomenprofilierung liefert Hinweise darauf, dass große nicht kodierende RNAs keine Proteine kodieren. Zelle 154, 240-251. doi: 10.1016/j.Zelle.2013.06.009
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Hsu, P. Y., Benfey, P. N. (2018). Klein aber oho: funktionelle Peptide, die von kleinen ORFs in Pflanzen kodiert werden. PROTEOMIK 18, 1700038. doi: 10.1002/pmic.201700038
Querverweis Volltext / Google Scholar
Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Hsu, P. Y., Wu, H.-Y. L., Rothfels, C. J., Ohler, U., et al. (2016). Superauflösende Ribosomenprofilierung zeigt nicht notierte Translationsereignisse in Arabidopsis. Prok. Natl. Acad. Sci. 113, E7126-E7135. doi: 10.1073/pnas.1614788113
CrossRef Volltext / Google Scholar
Ingolia, NT, Lareau, LF, Weissman, JS (2011). Die Ribosomenprofilierung von embryonalen Stammzellen von Mäusen zeigt die Komplexität und Dynamik von Säugetierproteomen. Zelle 147, 789-802. doi: 10.1016/j.Zelle.2011.10.002
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Ito, E. A., Katahira, I., Vicente, F. F., da, R., Pereira, L. F. P., Lopes, F. M. (2018). BASiNET-BiologicAl Sequences NETwork: eine Fallstudie zur codierenden und nicht-codierenden RNAs-Identifizierung. Nukleinsäuren Res. 46, e96-e96. doi: 10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093/bioinformatik/bty948
CrossRef Volltext / Google Scholar
Kearse, M. G., Wilusz, J. E. (2017). Nicht-AUG-Translation: ein neuer Start für die Proteinsynthese in Eukaryoten. Gene Dev. 31, 1717–1731. doi: 10.1101/gad.305250.117
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Zhang, Y., Ye, Z.-Q., Liu, X.-Q., Zhao, S.-Q., Wei, L., et al. (2007). CPC: Bewertung des Proteinkodierungspotentials von Transkripten mithilfe von Sequenzmerkmalen und Support Vector Machine. Nukleinsäuren Res. 35, W345-W349. doi: 10.1093/nar/gkm391
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Langmead, B., Salzberg, SL (2012). Schnelle gapped-lesen ausrichtung mit Bowtie 2. Nat. Methoden 9, 357-359. doi: 10.1038/nmeth.1923
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Meng, X., Chen, Q., Zhang, P., Chen, M. (2017). CircPro: ein integriertes Tool zur Identifizierung von circRNAs mit proteinkodierendem Potential. Bioinformatik 33, 3314-3316. doi: 10.1093/bioinformatik/btx446
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Michel, A. M., Baranov, P. V. (2013). Ribosomenprofilierung: Ein hochauflösender Monitor für die Proteinsynthese auf genomweiter Ebene: Ribosomenprofilierung. In: Wiley Interdiscip. Offenbarung, 4, 473-490. doi: 10.1002/wrna.1172
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Pamudurti, N. R., Bartok, O., Jens, M., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., et al. (2017). Translation von CircRNAs. Mol. Zelle 66, 9-21.e7. doi: 10.1016/j.molcel.2017.02.021
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Ponty, Y., Termier, M., Denise, A. (2006). GenRGenS: Software zur Erzeugung zufälliger genomischer Sequenzen und Strukturen. Bioinformatik 22, 1534-1535. doi: 10.1093/bioinformatik/btl113
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Rho, M., Tang, H., Ye, Y. (2010). FragGeneScan: Vorhersage von Genen in kurzen und fehleranfälligen Lesevorgängen. Nukleinsäuren Res. 38, e191-e191. doi: 10.1093/nar/gkq747
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Slavoff, S. A., Mitchell, A. J., Schwaid, A. G., Cabili, M. N., Ma, J., Levin, J. Z., et al. (2013). Peptidomic Entdeckung von kurzen offenen Leserahmen-codierten Peptiden in menschlichen Zellen. Nat. Chem. Biol. 9, 59–64. doi: 10.1038/nchembio.1120
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Spealman, P., Naik, A. W., May, G. E., Kuersten, S., Freeberg, L., Murphy, R. F., et al. (2018). Konservierte Nicht-AUG-uORFs, die durch eine neuartige Regressionsanalyse von Ribosomenprofildaten aufgedeckt wurden. Genome Res. 28, 214-222. Ursprungsbezeichnung: 10.1101/gr.221507.117
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Vicens, Q., Westhof, E. (2014). Biogenese zirkulärer RNAs. Zelle 159, 13-14. doi: 10.1016/j.Zelle.2014.09.005
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitation Willems, P., Ndah, E., Jonckheere, V., Stael, S., Sticker, A., Martens, L., et al. (2017). N-terminale Proteomik unterstützte die Profilerstellung der unerforschten Translationsinitiationslandschaft in Arabidopsis thaliana. Mol. Zelle. Proteomik 16, 1064-1080. Ursprungsbezeichnung: 10.1074/mcp.M116.066662
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Wu, J., Mao, X., Cai, T., Luo, J., Wei, L. (2006). KOBAS Server: eine webbasierte Plattform für automatisierte Annotation und Pfadidentifikation. Nukleinsäuren Res. 34, W720-W724. doi: 10.1093/nar/gkl167
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Yang, L., Fu, J., Zhou, Y. (2018). Zirkuläre RNAs und ihre aufkommende Rolle bei der Immunregulation. Front. Immunol. 9, 2977. doi: 10.3389/fimmu.2018.02977
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Yang, Y., Fan, X., Mao, M., Lied, X., Wu, P., Zhang, Y., et al. (2017). Umfangreiche Translation zirkulärer RNAs, angetrieben durch N6-Methyladenosin. Zelle Res. 27, 626-641. Ursprungsbezeichnung: 10.1038/cr.2017.31
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
Yu, G., Wang, L.-G., Han, Y., Er, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar