- Introduction
- ChREBP-Struktur und Regulation über die LID/Grace-Domänen
- Aktivierung von ChREBP durch Glucosemetaboliten
- ChREBP-Kofaktoren und Partner
- Rolle von ChREBP im Kohlenhydratstoffwechsel und der Hepatokinproduktion
- ChREBP als Regulator der hepatischen Fettsäuresynthese und VLDL-Sekretion
- Regulation des Fruktosestoffwechsels durch ChREBP in Leber und Darm
- Verordnung des BDK:PPM1K-Achse in der Leber
- ChREBP ist für die Glucose-vermittelte Regulation von FGF21 erforderlich
- Rolle von ChREBP im interorganellen Netzwerk, das die Energiehomöostase steuert
- Rolle von hepatischem ChREBP bei der Kontrolle des Insulinsensitivitätshaushalts
- Adipöses ChREBP verknüpft die Lipogenese mit der Insulinsensitivität
- Neuartige Wechselwirkung zwischen hormonsensitiver Lipase und ChREBP im Fettgewebe
- Fazit und zukünftige Richtungen
- Autorenbeiträge
- Finanzierung
- Interessenkonflikterklärung
Introduction
Der erhöhte Konsum von Einfachzuckern wie Saccharose und Maissirup mit hohem Fructosegehalt hat in den letzten Jahren zu einem erhöhten Risiko für Stoffwechselerkrankungen wie Fettleibigkeit, Dyslipidämie, Typ-2-Diabetes und / oder nichtalkoholische Fettlebererkrankungen (NAFLD) geführt. Die Leber ist das Hauptorgan, das für die Umwandlung überschüssiger Kohlenhydrate in Fett verantwortlich ist. Die resultierenden Triglyceride (TG) können in Lipoproteine mit sehr niedriger Dichte (VLDL) gepackt und entweder in den Kreislauf ausgeschieden, als Lipidtröpfchen gespeichert oder über den Beta-Oxidationsweg metabolisiert werden. Insulin, das als Reaktion auf erhöhten Blutzucker ausgeschieden wird, stimuliert die Expression von Genen der De-novo-Fettsäuresynthese (Lipogenese) durch den Transkriptionsfaktor Sterol regulatory Element binding Protein-1c (SREBP-1c) (Foretz et al., 1999). SREBP-1c wirkt in Synergie mit einem anderen Transkriptionsfaktor namens Carbohydrate Response Element Binding Protein (ChREBP), der die Reaktion auf Nahrungskohlenhydrate vermittelt. Die ChREBP-Proteinstruktur enthält eine Low Glucose inhibitory Domain (LID) und ein Glucose-Response Activation Conserved Element (GRACE), das sich in seinem N-Terminus befindet (Li et al., 2006). Die Aktivierung der GRACE-Domäne durch Glucosemetaboliten fördert die transkriptionelle Aktivität von ChREBP und die Bindung an eine hochkonservierte Sequenz namens Carbohydrate Response Element (ChoRE). ChoRE ist auf den Promotoren von ChREBP-Zielgenen vorhanden, die Schlüsselenzyme der De-novo-Lipogenese kodieren, einschließlich L-Pyruvatkinase (L-pk), einem geschwindigkeitsbegrenzenden Enzym bei der Glykolyse, Fettsäuresynthase (Fas), Acetyl-CoA-Carboxylase (Acc) und Stearoyl-CoA-Desaturase (Scd1) (Kawaguchi et al., 2001). Eine kürzlich durchgeführte Studie berichtete über die Interdependenz zwischen ChREBP (aktiviert durch Glukose) und SREBP-1c (aktiviert durch Insulin) für die vollständige Induktion der glykolytischen und lipogenen Genexpression in der Leber (Linden et al., 2018). Die virale Wiederherstellung der nuklearaktiven Form von SREBP-1c in der Leber von ChREBP-defizienten Mäusen (ChREBPKO) normalisierte die lipogene Genexpression, ohne die glykolytische Genexpression zu retten. Das Spiegelexperiment, bei dem die ChREBP-Expression in der Leber von SREBP-1c-Knockout-Mäusen induziert wurde, rettete die glykolytische Genexpression, aber überraschenderweise nicht die lipogene Genexpression, trotz der bekannten Rolle von ChREBP bei der Kontrolle von Genen der Fettsäuresynthese. Dennoch legt diese Studie die Bedeutung der doppelten Wirkung von ChREBP und SREBP-1c in den Kontrollgenen nahe, die an der Regulation der Fettsäuresynthese beteiligt sind (Linden et al., 2018).
ChREBP ist in der Leber stark angereichert und wurde als Hauptregulator des Fettstoffwechsels untersucht (Iizuka et al., 2004; Osorio et al., 2016). ChREBP wird auch signifikant in Pankreasinseln, Dünndarm, Skelettmuskel und in geringerem Maße in der Niere und im Gehirn exprimiert (siehe Richards et al., 2017 zur Überprüfung). Interessanterweise wurde eine andere Isoform von ChREBP, ChREBPß, die von einem alternativen ersten Exonpromotor stammt, erstmals im Fettgewebe identifiziert (Herman et al., 2012) und später in anderen Zelltypen beschrieben (siehe Abdul-Wahed et al., 2017 zur Überprüfung). Wie wir diskutieren werden, wird ChREBPß als konstitutiv aktive Isoform beschrieben. Es ist zu hoffen, dass zukünftige Arbeiten die jeweiligen Rollen von ChREBP und ChREBPß bei der Regulation des Glukose- und Lipidstoffwechsels untersuchen und ihre spezifischen und / oder überlappenden Ziele identifizieren werden.
ChREBP-Struktur und Regulation über die LID/Grace-Domänen
ChREBP gehört zur Mondo-Familie der bHLH/Zip-Transkriptionsfaktoren. Die N-Terminusdomäne (1-251 Reste) enthält zwei nukleare Exportsignale (NES) und ein nukleares Lokalisierungssignal (NLS), das die subzelluläre Lokalisation durch Interaktion mit der Chromosomenregionserhaltung 1 (CRM1) reguliert, die auch als Exportin 1 und / oder 14-3-3 Proteine (Sakiyama et al., 2008). Die C-Terminus-Region enthält eine Polypro-Domäne, eine bHLH / LZ-Domäne (660-737 Reste) und eine Leucin-Zipper-ähnliche Domäne (Zip-like, 807-847 Reste), die mit Co-Faktoren und DNA-Bindung assoziiert sind (Yamashita et al., 2001; Fukasawa et al., 2010; Ge et al., 2012). Lokalisierung und transkriptionelle Aktivierung von ChREBP werden durch die Nährstoffverfügbarkeit bestimmt. Die glukosevermittelte Regulation von ChREBP erfolgt meist auf der Ebene des Glucose-Sensing-Moduls (GSM) oder der Mondo-konservierten Region (MCR), die sich aus den LID- und den GRACE-Domänen zusammensetzt, wie in der Einleitung erwähnt (Abbildung 1A; In: Li et al., 2006; Singh und Irwin, 2016). Im Jahr 2012 haben Herman et al. (2012) beschrieben eine andere ChREBP-Isoform, ChREBPß, die von einem alternativen ersten Exonpromotor 1b zu Exon 2 transkribiert wird (Abbildung 1B). Dieses Transkript wird aus Exon 4 übersetzt und erzeugt ein kürzeres Protein mit 687 Aminosäuren (die ChREBP-Isoform in voller Länge, umbenannt in α, enthält 864 Aminosäuren, im Manuskript ChREBP genannt), in dem die beiden NES, NLS und die LID-Domäne fehlen. ChREBPß ist im weißen Fettgewebe in einer GLUT-4-abhängigen Weise hochaktiv und wird vorgeschlagen, direkt durch ChREBPa reguliert zu werden, da eine ChoRE-Sequenz im Exonpromotor 1β identifiziert wurde (Herman et al., 2012; Abbildung 1B). Die Regulation von ChREBPß durch ChREBPa legt die Existenz einer Feed-Forward-Schleife nahe, die möglicherweise die Reaktion auf Glukose unter hyperglykämischen Bedingungen verschlimmert. Der / die Regulationsmechanismus (e) der ChREBPß-Isoform und vor allem ihre spezifische Funktion müssen jedoch aufgeklärt werden.
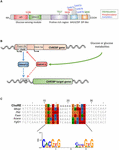
Abbildung 1. (A) Struktur des Kohlenhydrat-Antwortelement-Bindungsproteins α (ChREBPa). ChREBPa besteht aus 864 Aminosäuren und enthält mehrere regulatorische Domänen. Am N-Terminus enthält das Protein ein Glucose-Sensing-Modul, das aus der Low Glucose Inhibitory Domain (LID) und dem Glucose Activated Conserved Element (GRACE) besteht. Das Protein enthält auch eine polyprolin-reiche, eine bHLH / LZ und eine Leucin-Reißverschluss-ähnliche (Zip-like) Domäne am C-Terminus. Posttranslationale Modifikationen sind in ihren jeweiligen Resten Phosphorylierung (rot), Acetylierung (blau) und den kürzlich identifizierten O-Glcnacylierungen (grün) angegeben. (B) Genstruktur des ChREBP-Gens und Erzeugung der beiden ChREBP-Isoformen α und β. ChREBPß wird von einem alternativen ersten Exonpromotor 1b transkribiert. Dieses Transkript wird von Exon 4 translatiert, wodurch ein kürzeres Protein von 687 Aminosäuren erzeugt wird, in dem die beiden NES, die NLS- und die LID-Domäne fehlen. Es wurde vorgeschlagen, dass die ChREBPß-Isoform direkt durch ChREBPa reguliert wird, da eine ChoRE-Sequenz im Exonpromotor 1b identifiziert wurde. Ob sowohl chrebpß- als auch β-Isoformen an die ChoRE binden, ist derzeit nicht bekannt. Abbildung angepasst von Herman et al. (2012). (C) Mehrfachausrichtung von ChoRE-Konsensussequenzen, die in mehreren ChREBP-Zielgenpromotoren vorhanden sind. Die nukleotidbasierte Ausrichtung wird oben in der Abbildung zusammen mit der in Poungvarin et al. (2015). Das Logo, das der Konsensussequenz entspricht, die dieser bestimmten Ausrichtung zugeordnet ist, wird ebenfalls dargestellt.
Aktivierung von ChREBP durch Glucosemetaboliten
Unter Nüchternbedingungen ist die Glucagon-abhängige Aktivierung der Proteinkinase A (PKA) (Kawaguchi et al., 2002) phosphoryliert ChREBP an den Resten Ser196 und Thr666, was zur Bindung von ChREBP an das Protein 14-3-3 und dessen Retention im Cytosol führt (Kawaguchi et al., 2001, 2002; Davies et al., 2008). AMP-aktivierte Proteinkinase (AMPK), ein zentraler zellulärer Energiesensor, phosphoryliert auch ChREBP am Rest Ser568, was wiederum die Bindung von ChREBP an Promotoren seiner Zielgene verringert (Kawaguchi et al., 2002; Sato et al., 2016). Es wurde gezeigt, dass während des Fastens erzeugte Metaboliten wie AMP und Ketonkörper, die aus der Fettsäureoxidation hergestellt werden, eine allosterische hemmende Rolle spielen, indem sie die ChREBP- und 14-3-3-Proteinaffinität verändern, die Komplexstabilisierung verbessern und die zytosolische Retention begünstigen (Sakiyama et al., 2008; Nakagawa et al., 2013; Sato et al., 2016). Als Reaktion auf Kohlenhydrate wird ChREBP auf transkriptioneller, translationaler und posttranslationaler Ebene reguliert. Erhöhte Glukosekonzentrationen nach einer Mahlzeit fördern die Synthese von Zwischenmetaboliten wie Xylulose-5-phosphat (X5P), das ursprünglich als Aktivator der Proteinphosphatase 2A (PP2A) vorgeschlagen wurde (Kawaguchi et al., 2001; Kabashima et al., 2003). PP2A wurde zuvor beschrieben, um ChREBP am Ser196-Rest zu dephosphorylieren, was seine Translokation in den Kern ermöglicht, wo es in einer X5P- und PP2A-abhängigen Weise (auf Thr666 und Ser568) weiter dephosphoryliert wird. Dieses Modell wurde jedoch im Laufe der Jahre in Frage gestellt und andere Metaboliten wie Glucose-6-phosphat (G6P) wurden als potenzielle Aktivatoren der ChREBP-Translokation / -aktivität vorgeschlagen (Dentin et al., 2012). McFerrin et al. (2012) identifizierten ein mutmaßliches Motiv für die G6P-Bindung (253-SDTLFT-258) auf der GRACE-Domäne, die auch in MondoA, einem ChREBP / MondoB-Ortholog, konserviert ist (siehe Richards et al., 2017 zur Überprüfung). Nach dieser Hypothese könnte G6P eine allosterische Konformationsänderung fördern, die eine offene Konformation für ChREBP induziert, was die Interaktion mit Cofaktoren und die anschließende Translokation in den Kern erleichtert (McFerrin et al., 2012).
Innerhalb des Zellkerns kann ChREBP durch O-Glcnacylierung modifiziert werden, eine posttranslationale Modifikation, die vom Glukosestoffwechsel abhängt und als wichtig für die Transkriptionsaktivität von ChREBP identifiziert wurde (Guinez et al., 2010). Die O-Glcnacylierung erfolgt an Serin- und Threoninresten durch die Aktivität der O-GlcNAc-Transferase (OGT), einem Enzym, das N-Acetylglucosamin (GlcNAc) -Reste zu Zielproteinen hinzufügt, wodurch deren Aktivität, Stabilität und / oder subzelluläre Position modifiziert werden. Yang et al. (2017) enthüllten kürzlich mehrere durch O-Glcnacylierung modifizierte ChREBP-Rückstände. Mutationen dieser Reste an den bHLH / ZIP- und Dimerisierungs- und cytoplasmatischen Lokalisierungsdomänen (DCD) haben die Identifizierung von Thr517 und Ser839 als essentielle Stellen für die glucoseabhängige Aktivierung von ChREBP ermöglicht (Abbildung 1A). ChREBP kann auch durch Acetylierung über die Histon-Acetyltransferase-Aktivität von p300 modifiziert werden (Bricambert et al., 2010). Glucose-aktiviertes p300 acetyliert ChREBP auf Lys672 und erhöht seine Transkriptionsaktivität, indem es seine Rekrutierung für die ChoRE-Sequenz verbessert, deren optimale Konsensusbindungssequenz CAYGYCnnnnnCRCRTG ist (Abbildung 1C). Poungvarin et al. (2015) analysierten ChREBP-Bindungsstellen durch ChIP-Seq in Leber und weißem Fettgewebe von Mäusen, die mit einer kohlenhydratreichen, fettfreien Diät wieder gefüttert wurden. Sie berichteten, dass die ChREBP-Bindung in Signalwegen angereichert ist, die an Insulinsignalen, adhärenten Übergängen und Krebs beteiligt sind, was auf eine neuartige Beteiligung von ChREBP an der Tumorentstehung und dem Fortschreiten von Krebs hindeutet. Darüber hinaus berichtete eine kürzlich durchgeführte Studie über die Bedeutung von ChREBP beim hepatozellulären Karzinom (HCC) (Ribback et al., 2017). Die Autoren fanden heraus, dass die genetische Deletion von ChREBP (in ChREBPKO-Mäusen) die Hepatokarzinogenese beeinträchtigte, die durch die Proteinkinase B / Akt-Überexpression bei Mäusen verursacht wurde. Darüber hinaus führte die siRNA-vermittelte Hemmung von ChREBP in Maus- und / oder humanen HCC-Zellen zu einer verminderten Proliferation und Apoptose.
ChREBP-Kofaktoren und Partner
In den letzten Jahren wurden mehrere Kofaktoren und/oder Partner von ChREBP identifiziert (siehe Richards et al., 2017 zur Überprüfung). Max like Protein x (Mlx), ein bHLH/LZ-Transkriptionsfaktor, wurde erstmals als gemeinsamer Bindungspartner der Mondo-Familie identifiziert (Stoeckman et al., 2004). Die Dimerisierung von ChREBP mit Mlx ist sowohl für die nukleare Translokation als Reaktion auf Glucose als auch für die Bindung an ChoRE-Elemente erforderlich. Kernrezeptoren wie Hepatozyten-Kernfaktor 4α (HNF4a) und Farnesoid-X-Rezeptor (FXR) wurden ebenfalls als ChREBP-Partner beschrieben. HNF4a interagiert physikalisch mit ChREBP durch Bindung an die Direct Repeat-1 (DR-1) -Region auf dem Promotor von ChREBP-Zielgenen (Adamson et al., 2006; Meng et al., 2016). Darüber hinaus wurde gezeigt, dass die p300 / CBP-Transkriptions-Coaktivatorproteine den ChREBP / HNF4a-Komplex stabilisieren (Burke et al., 2009). Die transkriptionellen p300 / CBP-Coaktivatorproteine spielen eine zentrale Rolle bei der Koordination und Integration mehrerer signalabhängiger Ereignisse mit dem Transkriptionsapparat. Eine weitere Schlüsseleigenschaft von p300 / CBP ist das Vorhandensein von Histonacetyltransferase (HAT) -Aktivität, die p300 / CBP die Fähigkeit verleiht, die Chromatinaktivität durch Modulation nukleosomaler Histone zu beeinflussen. In menschlichen Hepatozyten löst die FXR-Bindung an den ChREBP-HNF4a-Komplex die Freisetzung von ChREBP aus CBP / p300 aus, was zur Rekrutierung der Histondeacetylase SMRT auf dem Lpk-Promotor führt und dadurch als Co-Repressor der ChREBP-Transkriptionsaktivität wirkt (Caron et al., 2013). Darüber hinaus modifiziert die CBP / p300-HAT-Aktivität ChREBP auf Lys 672, was zu seiner transkriptionellen Aktivierung als Reaktion auf Glucose führt (Bricambert et al., 2010).
Bricambert et al. (2018) identifizierten kürzlich den Histon-Demethylase-Pflanzenhomöodomain-Finger 2 (Phf2), der zur Familie der Histon-Lysin-Demethylase (KDM7) gehört, als neuartigen Cofaktor von ChREBP. Die Interaktion zwischen Phf2 und ChREBP verstärkt die transkriptionelle Aktivierung von ChREBP, indem H3K9-Methylmarkierungen auf dem Promotor seiner Zielgene gelöscht werden. Interessanterweise trägt die spezifische Co-Rekrutierung von Phf2 und ChREBP für den Promotor des Kernfaktors Erythroid 2 like 2 (Nrf2) zur schützenden Wirkung von Phf2 gegen erhöhte reaktive Sauerstoffspezies (ROS) und NAFLD-Progression im Zusammenhang mit Hyperglykämie bei (Bricambert et al., 2018).
Rolle von ChREBP im Kohlenhydratstoffwechsel und der Hepatokinproduktion
ChREBP als Regulator der hepatischen Fettsäuresynthese und VLDL-Sekretion
Die nichtalkoholische Fettlebererkrankung ist ein Kennzeichen des metabolischen Syndroms, und Studien am Menschen zeigen, dass die De-novo-Lipogenese bei Patienten mit NAFLD zu etwa 25% der gesamten Leberlipide beiträgt (Donnelly et al., 2005). In insulinresistenten Zuständen verstärken Hyperglykämie und Hyperinsulinämie die Lipogenese teilweise durch die Aktivierung von ChREBP und SREBP-1c. Die ChREBP-Hemmung in der Leber adipöser und insulinresistenter Ob / ob-Mäuse durch RNAi oder genetische Ablation führt zur Umkehrung der Lebersteatose (Dentin et al., 2006; Iizuka et al., 2006). Eine veränderte Sekretion von VLDL durch die Leber trägt ebenfalls zur Pathogenese von NAFLD bei. Das mikrosomale Triglyceridtransferprotein (MTTP) ist das Protein, das für die Assemblierung und Sekretion von Apolipoprotein-B-haltigen Lipoproteinen verantwortlich ist. Ein Mangel an MTTP bei Mäusen und Menschen verursacht Hypolipidämie und Fettleber. Die Regulation dieses Proteins wurde mit einigen hochkonservierten cis-Elementen in seinem Promotor in Verbindung gebracht, einschließlich kritischer positiver und negativer regulatorischer Domänen (Cuchel et al., 2013; Hussain et al., 2011). Kürzlich wurde ChREBP als potenzieller Regulator von MTTP hervorgehoben, da das Fehlen von funktionellem ChREBP in der Leber die Mttp-Expression und den VLDL-Aufbau und die Sekretion unterdrückt (Niwa et al., 2018). Da jedoch kein ChoRE am Mttp-Promotor eindeutig identifiziert werden konnte, sind weitere Analysen erforderlich, um den Mechanismus zu identifizieren, mit dem ChREBP Mttp reguliert.
Regulation des Fruktosestoffwechsels durch ChREBP in Leber und Darm
Der Zusammenhang zwischen ChREBP und Fruktosestoffwechsel wurde erstmals durch die phänotypische Analyse von ChREBP-Knockout-Mäusen (ChREBPKO-Mäusen) nachgewiesen. Es wurde berichtet, dass ChREBPKO-Mäuse innerhalb weniger Tage nach der Fütterung mit einer High-Fructose-Diät (HFrD) starben (Iizuka et al., 2004). Diese Hauptunverträglichkeit gegenüber Fructose wurde auf die Verringerung der Expression von Fructokinase und Triosekinase zurückgeführt, zwei Enzymen, die für den Fructosestoffwechsel erforderlich sind (Iizuka et al., 2004). In: Kim et al. (2016) berichtete später über die Bedeutung von ChREBP für die effiziente Umwandlung von Fructose in Glucose bei der Leber- und Ganzkörper-Fructose-Clearance, aber auch unter Einnahme von Fructose könnte ChREBP durch direkte Transaktivierung zur Hyperglykämie beitragen G6pc-Expression, ein Schlüsselgen der Glukoneogenese. Dieser Effekt könnte zu einem Teufelskreis führen, in dem der Fruktosekonsum die Glukoseproduktion durch ChREBP-Aktivität verstärkt (Kim et al., 2016). Im folgenden Jahr wurde die Studie von Zhang et al. (2017) berichteten, dass mit HFrD gefütterte ChREBPKO-Mäuse aufgrund einer Überaktivierung von endoplasmatischem Retikulum-Stress und einer CCAAT-Enhancer-bindenden proteinhomologen Protein (CHOP) -vermittelten Hepatozyten-Apoptose eine schwere Leberschädigung entwickeln. Die Apoptose in Hepatozyten bei diesen Mäusen war höchstwahrscheinlich mit einer erhöhten Cholesterinbiosynthese verbunden, da die Hemmung dieses Signalwegs über die HMG-CoA-Reduktase (HMGCR) oder die SREBP2-Hemmung die CHREBP2-Mäuse vor einer HFrD-induzierten Leberschädigung bewahrte. Ein Mangel an ChREBP wurde kürzlich auch mit einer Dysregulation des Saccharose- und Fructosestoffwechsels in Verbindung gebracht, die zu Zuckerintoleranz und Malabsorption bei Mäusen führte (Kato et al., 2018). Diese Effekte waren mit einer verminderten Expression von intestinaler Saccharose-Isomaltase (SI), die Saccharose in Glucose und Fructose verdaut, den Glucosetransportern 5 (Glut5) und 2 (Glut2) und dem Ketohexokinase (Khk) -Enzym, das die Fructolyse reguliert, verbunden (Abbildung 2). Eine Fehlregulation dieser Enzyme kann zur Ansammlung von unverdauter Saccharose und Fructose mit möglichen Auswirkungen auf die Zusammensetzung der Darmmikrobiota führen. Der Vergleich zwischen ChREBPKO- und leberspezifischen ChREBP-Knockout-Mäusen (ChREBPLiverKO), die mit HFrD gefüttert wurden, hatte zuvor gezeigt, dass ein hepatischer ChREBP-Mangel allein nicht zu einer Fruktoseintoleranz führt, sondern dass ein ChREBP-Mangel im Dünndarm höchstwahrscheinlich für die bei diesen Mäusen beobachtete Beeinträchtigung der Fruktosetoleranz verantwortlich ist (Kim et al., 2017). Insgesamt unterstreichen diese Studien die Bedeutung von ChREBP bei der Regulation des Fruktosestoffwechsels und unterstreichen die Notwendigkeit eines besseren Verständnisses seiner Rolle und Regulation im Dünndarm.
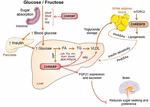
Abbildung 2. ChREBP reguliert multiples Signal- / Stoffwechselwege als Reaktion auf Glukose und Fruktose. ChREBP wird in mehreren Geweben exprimiert, einschließlich Darm, Leber und weißem Fettgewebe. In diesen Zelltypen wird ChREBP als Reaktion auf Glucose und / oder Fructose aktiviert und induziert ein spezifisches Genprogramm, wie in der Abbildung angegeben. Im Darm wurde die Stimulation der SI-, Glut5-, Glut2- und Ketohexokinase (Khk) -Expression durch ChREBP (entweder direkt oder indirekt) beschrieben, um die Saccharosetoleranz und die Fruktoseabsorption zu verbessern. In der Leber ist ChREBP ein Schlüsselmodulator der glykolytischen, lipogenen und mikrosomalen Triglyceridtransferprotein (Mttp) -Genexpression, wodurch sowohl die Fettsäureakkumulation als auch der VLDL-Export aus der Leber gesteuert werden. ChREBP reguliert auch die Produktion von Hepatokinen wie Fibroblastenwachstumsfaktor 21 (FGF21). Diese Leber-Hirn-Achse erweitert die ChREBP-Funktion der Leber von einem Leberregulator zu einem systemischen Modulator, der nicht nur die Substrathandhabung in der Leber, sondern auch die Nährstoffpräferenz beeinflusst. Die ChREBP-Aktivierung im weißen Fettgewebe ist mit einer verbesserten metabolischen Homöostase verbunden, indem schützende zirkulierende Signale erzeugt werden. Es wurde berichtet, dass eine neuartige Klasse von Säugetierlipiden, die durch eine verzweigte Esterbindung zwischen einer Fettsäure und einer Hydroxyfettsäure (Palmitinsäure-Hydroxylstearinsäure) gekennzeichnet ist, durch direkte und Inkretin-vermittelte Modulation der β-Zellfunktion positive Auswirkungen auf die Glucosehomöostase ausübt, verbesserte Fettglucoseaufnahme und reduzierte Entzündung. Interessanterweise wurde mTORC2 kürzlich als neuartiger Regulator der ChREBPß-Isoform in Fettzellen identifiziert.
Verordnung des BDK:PPM1K-Achse in der Leber
Der erste Schritt des Katabolismus verzweigtkettiger Aminosäuren (BCAAs) wird durch den verzweigtkettigen Ketosäuredehydrogenase (BCKDH) -Komplex reguliert, der durch zwei Enzyme, die verzweigtkettige Alpha-Ketosäuredehydrogenase-Kinase (BDK) und die Proteinphosphatase, Mg2 + / Mn22 + -1K (PPM1K), gesteuert wird. Weiß et al. (2018) assoziierten kürzlich ChREBP mit der Hochregulation von BDK und der Herunterregulation von PPM1K in der Leber und identifizierten ein konserviertes ChoRE-Motiv im Promotor dieser beiden Gene. Eine positive Korrelation zwischen der Expression von BDK und anderen typischen ChREBP-Zielgenen (Fasn, Pklr, ChREBPß) wurde in Lebern von Ratten beobachtet, die mit einer Diät mit hohem Glukose- oder Fruktosegehalt gefüttert wurden. Auf physiologischer Ebene führte eine Erhöhung des BDK:PPM1K-Verhältnisses zur Phosphorylierung und Aktivierung der ATP-Citratlyase (ACLY), wodurch die De-novo-Lipogenese stimuliert wurde. Diese Ergebnisse zeigen, dass BDK und PPM1K möglicherweise neuartige Lipogenese-aktivierende Gene sind, die durch ChREBPß reguliert werden. Aufgrund ihrer Rolle bei der Regulation des Lipid-, Glukose- und Aminosäurestoffwechsels könnten BDK und PPM1K in naher Zukunft als potenzielle therapeutische Ziele in der Leber in Betracht gezogen werden (White et al., 2018).
ChREBP ist für die Glucose-vermittelte Regulation von FGF21 erforderlich
ChREBP wurde kürzlich mit der Produktion und Sekretion von Hepatokinen wie dem Fibroblastenwachstumsfaktor 21 (FGF21) in Verbindung gebracht (Iizuka et al., 2009; Dushay et al., 2015, Iroz et al., 2017). FGF21 ist ein metabolisches Hormon, das von der Leber synthetisiert wird und mehrere positive Wirkungen in peripheren Geweben hat (Kharitonenkov et al., 2005; Badman et al., 2007; Markan et al., 2014). Bis vor kurzem wurde FGF21 als Fastenhormon angesehen, das die Fettsäureoxidation, Ketogenese und Lipolyse unter der Transkriptionskontrolle des Peroxisomen-Proliferator-aktivierten Rezeptors α (PPARa) verbessert (Inagaki et al., 2007). Eine lästige Pflicht am Fgf21-Promotor wurde zuvor sowohl bei Mäusen (-74 bis -52 bp) als auch bei Menschen (-380 bis -366 bp) identifiziert (Iizuka et al., 2009), aber funktionelle Studien fehlten bis vor kurzem. Es wurde berichtet, dass der Konsum von Glukose und Fruktose bei gesunden Probanden und Patienten mit metabolischem Syndrom zu einer raschen Erhöhung der FGF21-Spiegel führte (Dushay et al., 2015). Zusätzliche Studien berichteten auch über eine mechanistische Verbindung zwischen ChREBP-abgeleitetem FGF21 und Makronährstoffpräferenz über eine Leber-Hirn-Achse (Talukdar et al., 2016; von Holstein-Rathlou et al., 2016). Diese Leber-Hirn-Achse erweitert die ChREBP-Funktion von einem hepatischen Stoffwechselregulator zu einem systemischen Modulator und beeinflusst nicht nur die Handhabung von Lebersubstraten, sondern auch das globale Fütterungsverhalten (Abdul-Wahed et al., 2017).
Rolle von ChREBP im interorganellen Netzwerk, das die Energiehomöostase steuert
Rolle von hepatischem ChREBP bei der Kontrolle des Insulinsensitivitätshaushalts
Unser Labor berichtete zuvor, dass ChREBP als Schlüsselmodulator der hepatischen Fettsäurezusammensetzung und Insulinsensitivität im Zusammenhang mit nichtalkoholischen und alkoholischen Lebererkrankungen fungiert (siehe Abdul-Wahed et al., 2017 zur Überprüfung). Mäuse, die ChREBP überexprimierten, entwickelten eine größere Lebersteatose als Kontrollen, blieben aber interessanterweise frei von metabolischen Komplikationen und entwickelten keine Insulinresistenz. Die lipidomische Analyse hat gezeigt, dass die ChREBP-vermittelte Steatose mit einer Abnahme der gesättigten Fettsäuren und einer Zunahme der einfach ungesättigten Fettsäuren einhergeht, wobei letztere nachweislich mit ChREBP-vermittelten positiven Auswirkungen auf die Insulinsensitivität assoziiert sind (Benhamed et al., 2012). Diese Ergebnisse zeigen die Rolle von ChREBP bei der Lipidpartitionierung und legen nahe, dass bestimmte Lipidspezies, wenn sie an der richtigen Stelle und Zeit vorhanden sind, Signale auslösen können, die die Anpassung an metabolischen Stress modulieren (Benhamed et al., 2012; Bricambert et al., 2018). Interessanterweise haben Jois et al. (2017) schlugen auch eine schützende Rolle für hepatisches ChREBP in Bezug auf die Ganzkörperglukosehomöostase und die Insulinsensitivität vor. ChREBPLiverKO-Mäuse zeigen eine verschlechterte Glukosetoleranz, während sie vor Lebersteatose geschützt sind. Hepatische ChREBP-Deletion führte auch zu Veränderungen der Genexpression in weißen und braunen Fettgeweben, was auf eine Kommunikation zwischen den Geweben hindeutet. Der Beitrag von ChREBP zur Energiebilanz des gesamten Körpers kann daher auf seiner Regulation der Lipidspezies und / oder der Hepatokinproduktion beruhen, die zur Koordination der Energiehomöostase zwischen den Geweben beitragen (Jois et al., 2017).
Adipöses ChREBP verknüpft die Lipogenese mit der Insulinsensitivität
Eine gestörte Insulinsignalisierung im Fettgewebe ist ein kritisches Merkmal der Insulinresistenz. Studien haben berichtet, dass die ChREBP-Aktivierung im weißen Fettgewebe die metabolische Homöostase verbessern kann, indem schützende zirkulierende Signale erzeugt werden (Yore et al., 2014; Tang et al., 2016). Es wurde berichtet, dass eine Klasse von Säugetierlipiden, die durch eine verzweigte Esterbindung zwischen einer Fettsäure und einer Hydroxyfettsäure, Palmitinsäure-Hydroxylstearinsäure (PAHSA), gekennzeichnet ist, durch direkte und inkretinvermittelte Modulation der β-Zellfunktion positive Auswirkungen auf die Glukosehomöostase ausübt, Glukoseaufnahme und Verringerung der Entzündung (Yore et al., 2014). In ähnlicher Weise sind fettspezifische ChREBP-Knockout (ChREBPadiposeKO), die niedrige Lipogeneseraten im Fettgewebe aufweisen, insulinresistent mit beeinträchtigter Insulinwirkung in Leber, Muskel und weißem Fettgewebe sowohl unter Futter- als auch unter fettreichen Ernährungsbedingungen. ChREBPadiposeKO-Mäuse haben niedrigere Serumspiegel von PAHSAs, während eine PAHSA-Supplementierung, insbesondere das 9-PAHSA-Isomer, ChREBPadiposeKO vor Insulinresistenz und Fettgewebsentzündung rettet, was bestätigt, dass der Verlust von Fett-ChREBP ausreicht, um Insulinresistenz zu verursachen (Vijayakumar et al., 2017). Eine kürzlich durchgeführte Studie identifizierte das mechanistische Ziel von Rapamycin Complex 2 (mTORC2) als neuartigen Regulator von ChREBP (insbesondere der β-Isoform) in Fettzellen. Die spezifische Ablation von Rapamycin-unempfindlichem Begleiter von mTOR (Rictor) in reifen Adipozyten beeinträchtigte die insulinstimulierte Glukoseaufnahme im Fettgewebe, was zur Herunterregulierung von ChREBPß und zur Zielgenexpression führte, die an der Kontrolle der Lipogenese beteiligt waren (Tang et al., 2016). In Übereinstimmung mit einem wichtigen Fett–Leber-Übersprechen, das durch ChREBP vermittelt wird, sind diese Effekte mit einer Insulinresistenz in der Leber und einer verbesserten Glukoneogenese verbunden. Insgesamt unterstützen diese Studien eine wichtige Rolle des adipösen ChREBP bei der Auslösung insulinsensitiver Signale (Tang et al., 2016).
Neuartige Wechselwirkung zwischen hormonsensitiver Lipase und ChREBP im Fettgewebe
ChREBP wurde kürzlich als Partner des lipolytischen Enzyms hormonsensitive Lipase (HSL) im Fettgewebe identifiziert (Morigny et al., 2019). Es wurde gezeigt, dass der Knockdown von HSL in menschlichen Adipozyten und Mausfettgewebe die Insulinsensitivität erhöht und die Verlängerung des sehr langkettigen Fettsäureenzyms (Elovl6) induziert. Elov16 ist ein mikrosomales Enzym, das die Dehnung von C12-16 gesättigten und einfach ungesättigten Fettsäuren ChREBP-abhängig reguliert (Morigny et al., 2019). Auf mechanistischer Ebene beeinträchtigte die physikalische Interaktion zwischen HSL und ChREBP die nukleare Translokation von ChREBPa und die anschließende Induktion von ChREBPß und Zielgenen, insbesondere Elovl6 (Morigny et al., 2019). Diese Studie zeigt eine neuartige Regulation für ChREBP im Fettgewebe. Die Hemmung der Wechselwirkung zwischen HSL und ChREBP kann zu potenziellen therapeutischen Strategien zur Verbesserung der Insulinsensitivität in Fettzellen führen.
Fazit und zukünftige Richtungen
ChREBP ist jetzt ein etablierter Kohlenhydratsensor. Obwohl die meisten Studien seiner Bedeutung bei der Kontrolle der glykolytischen und lipogenen Signalwege gewidmet waren, haben jüngste Daten auch neue Beiträge von ChREBP in Hepatozyten und in Fettzellen aufgedeckt, wo es bei der Produktion von Hepatokinen und / oder Lipokinen, die ein Übersprechen zwischen Organen auslösen, eine Rolle spielen könnte. Wie diskutiert, neu identifizierte Kofaktoren (epigenetische Modifikatoren) und / oder Partner (adipöse HSL) in diesen Geweben können auch potenzielle therapeutische Strategien für NAFLD und / oder zur Verbesserung der systemischen Insulinsensitivität darstellen. Jüngste Studien haben auch die Bedeutung von ChREBP bei der Regulation des Fruktosestoffwechsels untermauert und die Notwendigkeit eines besseren Verständnisses seiner Rolle und Regulation im Dünndarm unterstrichen. Schließlich wird die Identifizierung spezifischer und / oder überlappender Ziele von ChREBPa und ChREBPß in Schlüsselzelltypen sowie die Bestimmung ihrer spezifischen Auswirkungen auf die Insulinsensitivität in den kommenden Jahren von besonderer Bedeutung sein.
Autorenbeiträge
Alle aufgeführten Autoren haben einen wesentlichen, direkten und intellektuellen Beitrag zum Werk geleistet und es zur Veröffentlichung freigegeben.
Finanzierung
Das Postic’s Lab (U1016-Institut Cochin) wird durch Zuschüsse des ChroME-Netzwerks (Marie Curie Skłodowska Action H2020-MSCA-ITN-2015-675610), der Foundation for the Medical Research (FRM) (DEQ20150331744) und des ANR-15-CE14-0026 unterstützt -Hepatokind.
Interessenkonflikterklärung
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.