Mit der Verwundung von gesundem Gewebe entfaltet sich ein vorhersehbarer Verlauf physiologischer Ereignisse. Dieses Fortschreiten kann in die Phasen Entzündung, Proliferation und Reifung unterteilt werden. Jede Phase ist durch die sequentielle Ausarbeitung charakteristischer Zytokine durch bestimmte Zellen gekennzeichnet. Sehen Sie die Bilder unten.
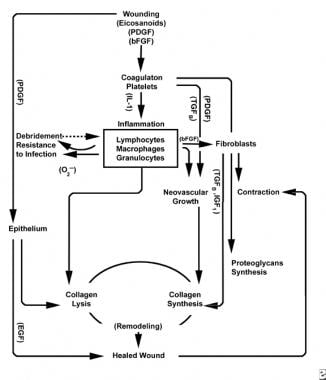 Schemata des Wundheilungsprozesses.
Schemata des Wundheilungsprozesses. 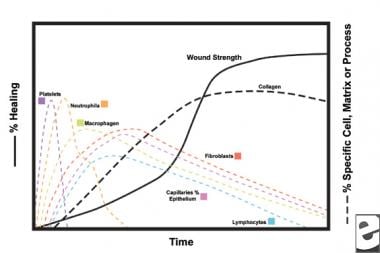 Zelluläre Eigenschaften des Wundheilungsprozesses.
Zelluläre Eigenschaften des Wundheilungsprozesses. Die Entzündungsphase
Die Entzündungsphase löst gleichzeitig hämostatische Mechanismen und Signalwege aus, die die klinisch erkennbaren Kardinalzeichen einer Entzündung erzeugen: Rubor (Rötung), Calor (Wärme), Tumor (Schwellung), Dolor (Schmerz) und functio laesa (Funktionsverlust).
Eine Verletzung des Gefäßgewebes löst die extrinsische Gerinnungskaskade aus, indem intrazelluläres Kalzium und Gewebefaktor freigesetzt werden, die den Faktor VII aktivieren. Dieser Pfropfen wirkt als Gitter für die Aggregation von Blutplättchen, dem häufigsten und „charakteristischen“ Zelltyp der frühen Entzündungsphase.
Blutplättchen enthalten eine Reihe entzündungsfördernder Substanzen wie Adenosindiphosphat, Gewebewachstumsfaktor Beta (TGF-ß) und von Blutplättchen abgeleitete Wachstumsfaktoren (PDGF). Diese Wachstumsfaktoren wirken auf umgebende Zellen und stimulieren die Chemotaxis von Neutrophilen, Monozyten und Fibroblasten in den Bereich der Verletzung.
Verletzte Gewebe katalysieren durch aktivierte Phospholipase A gleichzeitig Arachidonsäuren, um vasoaktive Prostaglandine und Thromboxan zu produzieren, die zusammen als Eicosanoide bekannt sind. Eicosanoide vermitteln Aktivität, die die Bildung von Blutplättchenpfropfen, die Gefäßpermeabilität und die zelluläre Chemotaxis beeinflusst, um die Wundheilung zu beeinflussen. Zum Beispiel vermittelt Thromboxan A2 Vasokonstriktion und Thrombozytenaggregation.
Nach anfänglicher Vasokonstriktion manifestieren sich die klassischen Entzündungszeichen durch erhöhte Gefäßpermeabilität. Rubor resultiert aus einer Vasodilatation, die durch Prostacyclin (PGI2), Prostaglandin A (PGA), Prostaglandin D (PGD) und Prostaglandin E (PGE) vermittelt wird. Tumor und Kalor entwickeln sich, wenn sich vaskuläre Endothellücken vergrößern, was den Austritt von Plasmaprotein und Flüssigkeit in den interstitiellen Raum ermöglicht. Diese Veränderungen werden durch PGE2 und Prostaglandin F2a (PGF2a) potenziert und ermöglichen das Eindringen von Entzündungszellen in den Bereich der Verletzung, einschließlich Zellen, die sich entwickeln. Dolor wird wahrgenommen, wenn PGI2, PGE und PGE2 auf periphere Nozizeptoren wirken.
Im zweiten Stadium der Entzündungsphase verdrängen Leukozyten Thrombozyten als dominanten Zelltyp, angezogen durch Chemotaxis. Weiße Blutkörperchen (WBCs) sind die vorherrschenden Zellen für die ersten 3 Tage nach der Verwundung; Ihre Anzahl erreicht ihren Höhepunkt nach ungefähr 48 Stunden. Polymorphonukleozyten (PMNs) beginnen als erste mit bakteriziden Aktivitäten unter Verwendung von Entzündungsmediatoren und Metaboliten freier Sauerstoffradikale. Eine normale Wundheilung kann jedoch ohne PMNs erfolgen. Ein weiterer Leukozyt, die Helfer-T–Zelle, arbeitet Interleukin-2 (IL-2) aus. IL-2 fördert die weitere Proliferation von T-Zellen, um die immunogene Reaktion auf Verletzungen zu verstärken.
Wenn PMN-Leukozyten nach 24-36 Stunden zu schwinden beginnen, gelangen zirkulierende Monozyten in die Wunde und reifen zu Gewebemakrophagen. Diese Zellen debriden die Wunde auf mikroskopischer Ebene und produzieren eine Vielzahl wichtiger Substanzen wie IL-1 und den basischen Fibroblastenwachstumsfaktor (bFGF). IL-1 stimuliert die Proliferation von Entzündungszellen und fördert die Angiogenese durch Endothelzellreplikation. bFGF ist ein chemotaktischer und mitogener Faktor für Fibroblasten und Endothelzellen. Im Gegensatz zu PMNs beeinträchtigt der Makrophagenmangel die Wundheilung stark, da Debridement, Fibroblastenproliferation und Angiogenese abnehmen.
Gegen Ende des Entzündungszyklus interagiert das sich entwickelnde Milieu von Eicosanoiden in der Wunde mit den vorhandenen Zelltypen, was zu einer Fibroblastensynthese von Kollagen und Grundsubstanz führt (aus erhöhtem Verhältnis von PGF2a zu PGE2). Darüber hinaus befinden sich die von Makrophagen abgeleiteten Wachstumsfaktoren jetzt auf optimalem Niveau und beeinflussen stark den Zustrom von Fibroblasten und dann Keratinozyten und Endothelzellen in die Wunde. Da mononukleäre Zellen weiterhin WBCs und Makrophagen ersetzen, beginnt die Proliferationsphase.
Die proliferative Phase
Zwei bis drei Tage nach der Verwundung wandern Fibroblasten von Wundrändern über die während der Entzündungsphase gebildete fibrinöse Matrix nach innen. Während der ersten Woche beginnen Fibroblasten, Glykosaminoglykane und Proteoglykane, die Grundsubstanz für Granulationsgewebe, sowie Kollagen als Reaktion auf Makrophagen-synthetisiertes bFGF und TGF-ß sowie PDGF zu produzieren.
Fibroblasten werden bald zum dominierenden Zelltyp und erreichen nach 1-2 Wochen ihren Höhepunkt. Sie erzeugen nicht nur Kollagenmoleküle, sondern auch Zytokine wie PDGF, TGF-ß , bFGF, Keratinozyten-Wachstumsfaktor und insulinähnlichen Wachstumsfaktor-1. Fibroblasten bauen auch Kollagenmoleküle zu Fasern zusammen, die vernetzt und in Bündeln organisiert sind. Kollagen ist der Hauptbestandteil des akuten Wundbindegewebes, wobei die Nettoproduktion für die nächsten 6 Wochen anhält. Der steigende Gehalt an Wundkollagen korreliert mit zunehmender Zugfestigkeit.
Keratinozyten und Endothelzellen vermehren sich während dieser Zeit ebenfalls und produzieren schließlich autokrine Wachstumsfaktoren, die ihr Wachstum aufrechterhalten. Die Endothelexpansion trägt zur Angiogenese bei, da intakte Gefäße Knospen im Granulationsgewebe erzeugen. Die Neovaskularisation erleichtert das Wachstum der fortschreitenden Fibroblastenlinie in die Wunde und versorgt sie mit den notwendigen Nährstoffen und Zytokinen.
Der Abbau des Fibringerinnsels und der provisorischen Matrix geht einher mit der Ablagerung von Granulationsgewebe (Grundsubstanz, Kollagen, Kapillaren), das sich bis zur Wundbedeckung fortsetzt. Abnehmende Hyaluronsäure (in der Grundsubstanz) und steigende Chondroitinsulfatspiegel verlangsamen die Fibroblastenmigration und -proliferation, während sie die Fibroblastendifferenzierung induzieren und in die Reifungsphase der Wundheilung übergehen.
Die Reifungsphase
In den ersten 6 Wochen dominiert die Produktion von neuem Kollagen den Wundheilungsprozess und lagert sich zufällig im akuten Wundgranulationsgewebe ab. Wenn die Wunde reift, wird Kollagen in eine besser organisierte Struktur mit erhöhter Zugfestigkeit umgewandelt. Allmählich ersetzt Kollagen Typ I Typ III, bis das normale Hautverhältnis von 4: 1 erreicht ist. Mit fortschreitender Remodellierung erreicht die Matrix-Metalloproteinase-Kollagenolyse einen Steady State mit der Kollagensynthese. Zugfestigkeit Plateaus bei 80% der ursprünglichen Festigkeit etwa 1 Jahr postinjury.
Aufgrund dieser Aktivität wandern Epithelzellen vom Wundrand weiter nach innen, bis der Defekt bedeckt ist. An diesem Punkt induziert die Kontakthemmung die Umwandlung von Fibroblasten in Myofibroblasten, die kontraktile Aktinfasern enthalten. Es folgt eine Wundkontraktion, bei der das verletzte Gewebevolumen durch neues Gewebe ersetzt wird, obwohl die genaue Rolle des Myofibroblasten nicht vollständig geklärt ist.
Abschreckungsmittel zur Wundheilung
Akute Wunden durchlaufen im Allgemeinen einen geordneten und rechtzeitigen Reparationsprozess, der zu einer dauerhaften Wiederherstellung der anatomischen und funktionellen Integrität führt. Verschiedene physiologische und mechanische Faktoren können jedoch die Heilungsreaktion beeinträchtigen, was zu einer chronischen Wunde führt, die nicht den üblichen schrittweisen Verlauf durchläuft. Lokale Infektionen, Hypoxie, Traumata, Fremdkörper oder systemische Probleme wie Diabetes mellitus, Mangelernährung, Immunschwäche oder Medikamente sind am häufigsten dafür verantwortlich.
Alle Wunden sind kontaminiert, widerstehen aber am erfolgreichsten invasiven Infektionen. Wenn die Konzentration 100.000 (105) Organismen pro Gramm Gewebe überschreitet oder das Immunsystem beeinträchtigt wird, kommt es häufig zu Infektionen. Zellulitis verlängert die Entzündungsphase, indem sie ein hohes Maß an proinflammatorischen Zytokinen und Gewebeproteasen aufrechterhält, die Granulationsgewebe und Gewebewachstumsfaktoren abbauen, und indem sie die Kollagenablagerung verzögert.
Debridement (chirurgisch, enzymatisch und / oder durch Verbandwechsel) und Antibiotika sind die Hauptstützen der Antibiotikabehandlung. Debridement entfernt devitalisiertes Gewebe, das eine Quelle von Endotoxinen sein kann, die die Migration von Fibroblasten und Keratinozyten in die Wunde hemmen. Fremdkörper können ebenfalls entfernt werden müssen, da das Vorhandensein einer Seidennaht die Anzahl der Bakterien reduziert, die erforderlich sind, um eine Infektion um das 10.000-fache auszulösen. (Für eine detaillierte Beschreibung der Technik, siehe Medscape Referenzartikel Wunde Fremdkörperentfernung.)
Zelluläre Hypoxie verzögert die Wundheilung auf verschiedene Weise. Die Vernetzung der Kollagenfibrillen erfordert Sauerstoff, um Prolin und Lysin zu hydroxylieren, und versagt, wenn der Gewebedruck unter 40 mm Hg liegt. Die bakterizide Wirksamkeit der oxidativen Phosphorylierung von Leukozyten leidet auch in einer hypoxischen Umgebung, wodurch die Infektionsschwelle verringert wird. Maßnahmen zur Verbesserung der Sauerstoffzufuhr hängen von der Ätiologie ab. Der Tabakkonsum, der eine Vasokonstriktion verursacht und die Thrombozytenadhäsion erhöht, sollte gestoppt werden. Bei peripheren Gefäßerkrankungen kann eine Angioplastie oder eine arterielle Bypass-Transplantation erforderlich sein. Zusätzliche Maßnahmen zur Verbesserung der systemischen Perfusion bei Herzinsuffizienz können angezeigt sein. Ein Hämatokritwert von weniger als 15% sollte nach Bedarf behandelt und die Euvolämie wiederhergestellt werden. Venöse Stase oder lymphatische Insuffizienz kann mit Kompressionsstrümpfen verbessert werden.
Systemische Erkrankungen können die Wundheilung dramatisch verlängern oder unterbrechen. Glykosylierung bei Diabetes mellitus beeinträchtigt die neutrophile und Makrophagen-Phagozytose von Bakterien und verlängert die Entzündungsphase. Die proliferative Phase ist auch bei derselben Krankheit langwierig, da Erythrozyten weniger biegsam werden und weniger in der Lage sind, der Wunde Sauerstoff für den Gewebestoffwechsel und die Kollagensynthese zuzuführen.
Mangelernährung führt zu verminderter Fibroblastenproliferation, beeinträchtigter Neovaskularisation und verminderter zellulärer und humoraler Immunität. Wunden stellen erhöhte metabolische Anforderungen, insbesondere im Granulationsgewebe. Aminosäuren wie Methionin, Prolin, Glycin und Lysin sind essentiell für die normale Zellfunktion und die Reparatur von Hautwunden. Fettsäuren sind kritische Bestandteile von Zellmembranen und das Substrat für die Eicosanoide, die den Entzündungsprozess vermitteln. Essentielle Fettsäuren Linolensäure und Linolsäure müssen über die Nahrung zugeführt werden, da der menschliche Körper nicht in der Lage ist, diese Moleküle de novo zu synthetisieren.
Für den Zellstoffwechsel müssen ausreichende Vitamine und Mineralstoffe zur Verfügung stehen, die als zelluläre Signale und Cofaktoren wirken. Vitamin C (Ascorbinsäure) und Eisen werden für die Hydroxylierung von Lysin und Prolin benötigt, die die Tripelhelixstruktur von Kollagen vernetzen und stabilisieren; Kupfer spielt auch eine Rolle bei der Stabilisierung von Kollagen. Vitamin A (Retinsäure) spielt eine wichtige Rolle bei der Modulation der Kollagenproduktion und des Kollagenabbaus und ist besonders wichtig für die Epithelisierung. Vitamin E (Alpha-Tocopherol), ein starkes Antioxidans, scheint die Haut- und Knochenheilung bei Tieren zu beschleunigen, und eine Supplementierung kann beim Menschen eine Rolle spielen. Spurenmetall, insbesondere Zink, Mangel ist auch mit einer schlechten Wundheilung verbunden; Dies sollte gegebenenfalls wieder aufgefüllt werden.
Ovid schrieb: „Medikamente heilen manchmal, manchmal töten sie.“ Dies gilt sicherlich für die Wundheilung. Kortikosteroide stumpfen die Prozesse der gesamten Entzündungsphase ab. Vitamin A (topisch oder 25.000 IE / tag oral) mildert die nachteiligen heilenden Wirkungen von Kortikosteroiden, aber Hepatotoxizität kann bei längerem Gebrauch auftreten (dh > 1 mo). Nichtsteroidale entzündungshemmende Medikamente (NSAIDs) stören auch den Arachidonsäurestoffwechsel und damit die Wundheilung. Zusätzlich hemmen NSAIDs die Thrombozytenfunktion, einen der frühesten Prozesse in der Entzündungsphase.
Eine Studie von Sutcliffe et al schlug vor, dass die Hochregulierung des Gap-Junction-Proteins Connexin bei chronischen Wunden üblich ist. Untersuchung von Connexin in drei Arten von Wunden – venöses Bein, diabetischer Fuß und Dekubitus — Die Forscher fanden heraus, dass jede Art von Wunde eine Hochregulierung von epidermalem Connexin 43, Connexin 26 und Connexin 30 sowie dermal Connexin 43 zeigte.