Introdução
Membro-Cinto de Distrofias Musculares (LGMDs) incluem um heterogéneo grupo de transtornos caracterizados pela progressiva a desolação e a fraqueza proximal do membro-calças músculos (1, 2). Os LGMDs apresentam variabilidade inter e intra-ramiliar, variando de formas muito ligeiras, a fenotipos graves, precoces e rapidamente progressivos (2). LGMDs pode ser classificado como autossômico dominante (LGMD1) ou recessivo (LGMD2). O primeiro grupo geralmente tem uma idade adulta de início e não são muito comuns (<10% de todos os LGMDs), enquanto os últimos são mais frequentes (1:15000) (1). Entre as formas recessivas, o LGMD2A (ou calpainopatia) é o LGMD mais comum em todo o mundo, afetando aproximadamente 30% de todos os casos LGMDs (3). As assinaturas clínicas da doença incluem andar em bicos de pés, andar cambaleante, dificuldade em correr e subir escadas, ondulação escapular. Contraturas articulares, encurtamento do tendão de Aquiles, escoliose são comumente observados, enquanto os músculos faciais e do pescoço não são afetados (4). Uma Hiperckemia assintomática (5-80 vezes os níveis normais de CPK) em doentes jovens é considerada uma fase pré-clínica da doença e pode persistir durante vários anos (1, 5). A idade de início da fraqueza muscular geralmente ocorre aos 15 anos, embora possa ocorrer em idades anteriores (<12 anos) ou posteriores (>30 anos). A progressão da doença pode levar à perda de ambulação, insuficiência respiratória e redução da capacidade vital pulmonar nos estágios avançados (6). O envolvimento cardíaco (alterações do ritmo cardíaco, perturbações da condução cardíaca, disfunção de ejecção ventricular esquerda) é apenas ocasionalmente notificado (6, 7).
o diagnóstico de calpainopatia é confirmado pela detecção de mutações patogénicas no CAPN3 (15q15.1) (4), codificando diferentes transcrições alternativamente splicadas. No entanto, a transcrição completa é expressa principalmente no tecido muscular (8). A proteína codificada (CAPN3) é um membro da família das proteases cisteínicas não-lisossômicas dependentes de Ca++. No músculo, CAPN3 participa na” remodelação sarcomere”, que é essencial para a adaptação muscular e crescimento em resposta às demandas funcionais e metabólicas. Até à data, foram descritas mais de 490 mutações patogénicas em todo o CAPN3, a maioria das quais são alterações de nucleótidos únicos (4). Mutações no CAPN3 têm sido associadas com anormalidades mitocondriais, falha do crescimento, aumento do estresse oxidativo e desorganização sarcomérica que, ao todo, contribuem para tornar o músculo incapaz de manter cargas, causando assim degeneração miofiber e desperdício muscular (8). O diagnóstico da calpainopatia pode ser um desafio devido à heterogeneidade genética e à não especificidade do padrão clínico e instrumental. De facto, a distribuição da fraqueza/atrofia muscular, hipertrofia/pseudo-hipertrofia e contracturas dos tendões são frequentemente partilhadas com outros LGMDs ou perturbações neuromusculares. O padrão de biópsia muscular na calpainopatia é geralmente não específico também, variando de anomalias musculares ligeiras a alterações distróficas graves. Além disso, os marcadores imunohistoquímicos/bioquímicos não são geralmente fiáveis.: o sinal de calpain pode ser normal mesmo na presença de uma proteína não funcional, e vice-versa pode ser reduzido mesmo em outras distrofias musculares diferentes da calpainopatia.Por conseguinte, recomenda-se a realização de um diagnóstico diferencial a fim de obter resultados precisos e fiáveis. Para este efeito, foram desenvolvidas abordagens de testes genéticos moleculares para confirmar o diagnóstico de calpainopatia, incluindo painel multigeno, genoma extenso (sequenciação exome/genoma) e análise de um único gene (sequenciação direta) (9). A implementação da sequenciação de próxima geração (NGS) foi útil para gerar dados informativos a serem aplicados para fins diagnósticos, preditivos ou terapêuticos (10-12). Os painéis de genes NGS são baseados na análise de um conjunto de genes associados a uma doença específica ou um grupo de distúrbios relacionados, que são caracterizados pela heterogeneidade genética e fenotípica. Painéis NGS também podem ser úteis para detectar fenótipos complexos ou misturados, que resultam da herança de mais de um defeito genético dos pais (13). Em geral, os pacientes que apresentam fenótipos complexos, que são negativos para as mutações conhecidas em painéis de genes diagnósticos personalizados e que requerem um diagnóstico diferencial amplo, são elegíveis para o Exoma completo ou sequenciamento do genoma. O sequenciamento completo do exome / genoma são abordagens mais dispendiosas em termos de gestão de Dados, Interpretação e custos analíticos, mas aumentam a probabilidade de fornecer um diagnóstico molecular a uma suspeita de doença genética (13). No que diz respeito aos LGMDs, são altamente recomendados painéis específicos de NGS, para garantir taxas de diagnóstico elevadas, cobertura óptima, sensibilidade e especificidade dos testes clínicos (9). Os painéis NGS representam, portanto, um dos melhores sistemas para facilitar o diagnóstico diferencial, identificar novas mutações causativas e clarificar correlações genótipo-fenótipo (14, 15).
neste manuscrito, o caso de um paciente afetado por LGMD2A é relatado, para que a aplicação do SNGN painel, permitiu não só para confirmar o diagnóstico de calpainopathy (mutações no CAPN3), mas também para identificar um adicional de, romance mutação no gene LMNA associados com cardiomiopatia dilatada. Tendo em conta estes resultados, a análise foi alargada aos membros da família do proband para fornecer uma interpretação mais abrangente dos dados analíticos em relação aos fenótipos patológicos.
Apresentação Do Caso
caracterização Clínica de Proband e parentes
o aparecimento da doença no proband foi referido aos 10 anos de idade, quando ela relata a presença de hipertrofia do vitelo com andar na ponta dos pés, dificuldades em correr, escadas de escalada e de pé. Os sintomas mostraram um agravamento lento mas progressivo, com subsequente envolvimento do membro superior proximal. Aos 13 anos de idade, níveis elevados de CPK (~8.000 UI/L) foram incidentalmente descobertos, nunca associados com mioglobinúria. Sucessivamente, o paciente passou por várias investigações neuromusculares em vários centros especializados. Duas biópsias musculares foram realizadas, mostrando um quadro distrófico clássico (fibras hipertróficas / atroficas, núcleos internos, fibras necróticas, aumento do tecido conjuntivo) com características não específicas. Como esperado, os estudos de imunoquímica e imunoblot foram inconclusivos indicando sinais anormais / reduzidos de α, β, γ sarcoglicano, β-distroglicano e distrofina apenas em fibras necróticas (imunoquímica repetida resultou normal, também para a laminina).; imunoblots para distrofina e calpain revelaram sinais normais. No entanto, o imunoblot calpain-3 é conhecido por não ser completamente sensível para o diagnóstico de LGMD2A, uma vez que 20-30% dos casos apresentam uma quantidade normal de proteína (1, 16-18). Devido à apresentação clínica (hiperckmia, hipertrofia de vitelos/pseudo-hipertrofia), as distrofinopatias foram, em primeiro lugar, excluídas. Além disso, a análise dos genes DMD (Xp21), FKRP (19T13, 32) e DYSF (2p13, 2) não revelou mutações patogénicas. O paciente chegou à nossa observação aos 30 anos, apresentando envolvimento axial e cinta tanto no membro superior quanto no inferior, com significativa marcha e escápulas aladas. Nos membros inferiores, a fraqueza era proeminente nos quadriceps e músculos glutei que eram significativamente ipo-atróficos, juntamente com a evidência de “aparentemente hipertróficos” (pseudo-hipertrofia) músculos da barriga da perna (Figura 1). Não foram observadas cãibras, mialgias e ondulações. Os exames pneumológicos completos (espirometria e polissonografia) e cardiológicos (ecografia, monitorização de Holter de 24-h, IRM cardíaca, inspecção física cardiológica completa com história clínica específica e análise de reflexos cardiovasculares) não revelaram qualquer anomalia significativa. Também excluímos a deficiência de maltase ácida (ensaios de actividade enzimática normal em linfócitos/leucócitos). Músculo Imagens de Ressonância Magnética (MRI), mostrou um proeminente envolvimento da cintura escapular, músculos paravertebrais posterior do compartimento músculos da coxa (com relativa preservação da sartorius e gracilis músculos) e da perna (em particular, gastrocnêmio medial e sóleo) (Figura 2). Este padrão de envolvimento já foi notificado na literatura relativa ao quadro de IRM na calpainopatia (19, 20). Além disso, o estudo de IRM mostrou que os músculos dos bezerros foram efetivamente atróficos e caracterizados por infiltração de gordura, causando um aumento muscular”.”A apresentação clínica, a idade do início, a taxa de progressão, a distribuição da fraqueza muscular e os achados de IRM no proband foram totalmente consistentes com um diagnóstico de LGMD2A extensivamente descrito na literatura (4, 21, 22).
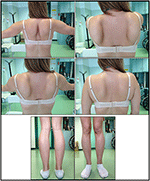
Figura 1. Características clínicas Proband: escápulas aladas e combinação de atrofia da coxa e pseudo-hipertrofia da perna.
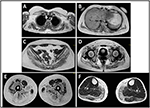
Figura 2. IRM muscular de Proband: o envolvimento de membros superiores cinto e músculos paravertebrais (A,B), o envolvimento dos membros inferiores cinto e compartimento posterior da coxa (C–E, principalmente os glúteos), com relativa preservação da sartorius e gracilis, e o envolvimento do compartimento posterior da perna (F, principalmente gastrocnêmio medial/sóleo).
a avaliação dos membros da família do proband não revelou qualquer história de doença neuromuscular e nenhuma evidência de consanguinidade entre os pais. No entanto, a mãe do proband, aos 55 anos, apresentou 2 episódios sincopais para os quais foi diagnosticada bradicardia idiopática (até 40 bpm por noite). Extensas investigações cardiológicas na mãe do proband diagnosticaram um bloqueio auriculoventricular sintomático (AV) após a ocorrência de vários episódios de síncope/lipotimia. A análise mostrou a presença de um bloco AV básico de primeiro grau, com vários períodos de bradicardia grave, principalmente noturna e muitas vezes sintomática, associada a algumas fases de bloqueio AV de segundo grau, tanto tipo 1 Quanto 2,e algumas fases de dissociação AV completa noturna. A ecocardiografia e o teste ergométrico não revelaram alterações significativas, com excepção da patente foramen ovale. A mãe não apresentou qualquer outro sintoma clínico, condição predisponente ou factor de risco. Dado o seu quadro clínico, a mãe do proband foi implantada com PMK. Além disso, a história da família revelou que a avó e o bisavô do proband também foram implantados com PMK aos 55 e 30 anos, respectivamente. Além disso, o irmão da avó do proband morreu aos 51 anos por cardiomiopatia dilatada grave. Exames cardiológicos também foram realizados no Pai e no tio materno do proband que resultou completamente não afetado.O presente estudo foi aprovado pelo Comitê de Ética da Fundação Santa Lúcia e realizado de acordo com a Declaração de Helsinque. Todos os participantes deram o consentimento informado e assinado para a análise genética e, a este respeito, também deram o consentimento para a publicação deste relatório de caso.O ADN genómico foi extraído do sangue periférico (400 µL) utilizando o Kit de extracção de adn do sangue de MagPurix e o sistema de extracção automática de MagPurix (Resnova) de acordo com as instruções do fabricante. As amostras foram sequenciadas usando o sistema PGM iônico e Ampliseq painel personalizado de alta especificidade (Thermo Fisher Scientific). O tamanho do painel era de 129.O painel incluiu 18 genes, que foram seleccionados com base na literatura científica, Generevews (www.ncbi.nlm.nih.gov/books/NBK1116/) e frequência das variantes patogénicas na população em geral. O quadro 1 resume uma descrição pormenorizada do painel NGS.
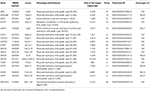
Quadro 1. Painel NGS personalizado utilizado para diagnóstico de LMGMD.
a construção de Bibliotecas foi realizada por Kits de biblioteca Ion AmpliSeq™ 2.0. Aproximadamente 10 ng / µl de DNA inicial foram utilizados para reações de PCR multiplex. Sucessivamente, duas etapas de purificação (usando AMPure XP, Beckman Coulter) foram realizadas para remover contaminantes indesejados e uma PCR final foi realizada. As fases de amplificação e enriquecimento do modelo foram realizadas por Ion PGM Hi-Q OT2 kit-400, Sistema de OneTouch 2 iónico e OneTouch ES iónicos (Thermo Fisher Scientific). Samples were processed by Ion PGM Hi-Q Sequencing Kit (400 bp, Thermo Fisher Scientific) and run on Ion 316 Chip v2 (850 flows required) and Ion PGM Sequencer (Thermo Fisher Scientific). Os resultados foram analisados usando o repórter iônico 4.6 (Thermo Fisher Scientific) e o visualizador integrado do genoma (IGV). A interpretação das variantes genéticas foi conduzida pela Human Gene Mutation Database (HGMD), Leiden Open Variation Database (LOVD), ClinVar and ExAC. O efeito funcional das variantes detectadas foi avaliado por ferramentas de previsão bioinformáticas, incluindo o provador de mutação, Varsome, SIFT, PolyPhen 2, SMART, Human Splicing Finder (HSF). Sequenciamento direto (Bigdye Terminator v3.1, BigDyeX Terminator e ABI3130, applied Biosystems) foi realizada para confirmar as variantes genéticas e a seqüência genômica de codificação regiões com cobertura <20X (LMNA, Chr1:156106052-156106076; DYSF, Chr2:71753352-71753502, Chr2:71776385-71776640; SGCB, Chr4:52904225-52904560; SGCA, Chr17:48243242-48243570).
Results
The NGS analysis of the proband revealed 3 heterozygous variants (Supplementary Figure 1). Duas variantes foram localizados no CAPN3, nomeadamente NM_000070.2 (CAPN3): c.550delA (p.Thr184Argfs) e c.1813G>C (p.Val605Leu) no exon 4 e 16, respectivamente. A primeira é a eliminação de um único nucleótido e é a variante patogénica mais comum (75% dos casos) nos Países Europeus (1). Como esperado, a análise Bioinformática classificou o C. 550delA (rs80338800) como uma variante de perda de função (P. Thr184Argfs), causando uma mudança de frame de leitura aberta. As ferramentas preditivas (provador de mutação, HSF, Varsome, PolyPhen 2, SIFT) descreveram a variante como deletéria. Além disso, SMART revelou que o produto proteico alterado não possui o domínio calpain 3 e os três motivos “EF-hands”. O primeiro domínio está envolvido nas vias de sinalização de Calpain, enquanto as mãos EF são essenciais para a ativação da proteína dependente de Ca++(4).
Relacionadas c.1813G>C, é um romance missense variante (p.Val605Leu) que está previsto para ser prejudiciais (por Mutação Provador, HSF, Varsome, PolyPhen 2, PENEIRE a) por causa de uma potencial alteração de splicing. Esta variante não é anotada na literatura ou nas bases de dados online e não foi encontrada em 200 sujeitos de controlo. Infelizmente, a análise por SMART tool não produziu resultados significativos, uma vez que a variante está localizada dentro de um domínio não catalogado. A análise de segregação de CAPN3 mostrou que a mãe e o tio materno foram ambos heterozigotos para c.550delA, enquanto o pai era portador de c.1813G>C.
além disso, o NGS análise sobre o proband revelou a presença de uma nova variante em LMNA, nomeadamente NM_170707 (LMNA): c.550>T (p.Gln184*). A variante sobre-mencionada foi prevista para ser uma variante nula, ainda não foi descrita na literatura ou entre bases de dados online e não foi encontrada em 200 sujeitos de controle. Ferramentas preditivas (provador de mutação, HSF, Vansome, PolyPhen 2, SIFT) descreveram um efeito patogénico desta variante sobre o produto proteico. SMART tool relatou que a proteína LMNA alterada carece do domínio do filamento, que é essencial para manter sua estrutura e função. A análise da segregação destacou a presença do C.550c >t variante apenas na mãe, sugerindo uma possível associação com a sintomatologia Cardíaca e a história familiar de doença.
de Acordo com os critérios estabelecidos pelo Colégio Americano de Genética Médica (ACMG) as Normas e Diretrizes (23), c.1813G>C (CAPN3) pode ser descrito como um provável patogênicos variante considerando que ele está localizado em um domínio críticas sem benigna variação (PM1); ela está ausente na ExAc, GnomAD, e 1000 Genoma bancos de dados do Navegador (PM2); considerando o modelo recessivo de herança, foi detectado em trans com uma variante patogênica (PM3); múltiplas linhas de evidência computacional suportam um efeito deletério no gene ou produto genético (PP3). Sobre a classificação clínica da c.550>T (LMNA) por ACMG, ele pode ser designado como um patogênicos variante, uma vez que é uma variante nulo, causando perda de função (p.Gln184*) em LMNA (PVS1); ela está ausente na ExAc, GnomAD, e 1000 Genoma bancos de dados do Navegador (PM2); ele está localizado em um domínio críticas sem benigna variação (PM1); múltiplas linhas de evidência computacional suportam um efeito deletério no gene ou produto genético (PP3).
Conclusões
Este relato de caso apresentado um paciente afetado por LGMD, que foi encontrado para ser portadora de mutações não só em CAPN3 (c.550delA e c.1813G>C), mas também em LMNA (c.550>T). Estes resultados explicam o fenótipo neuromuscular do proband por causa das mutações CAPN3 e realçam um risco potencial de distúrbios cardiovasculares devido à presença da variante na LMNA e sua história familiar positiva. Tendo em conta estes resultados, os membros da família foram submetidos à análise da segregação. Em particular, a mãe resultou em ser portadora de CAPN3_c. 550delA e LMNA_c.550C>T, respectivamente. A mãe pode ser considerada como um portador saudável para a mutação patogénica CAPN3_c.550delA associada à calpainopatia. No entanto, a presença de uma nova variante na LMNA pode explicar a sua patologia cardiovascular (bradicardia e episódios sincopais). A ausência de sintomatologia neuromuscular na mãe e o quadro clínico peculiar do proband excluíram uma possível associação de LMNA_c.550C> T com fenótipo neuromuscular, especialmente no que respeita à distrofia Muscular Emery-Dreifuss (EMD). Na verdade, EMD afeta especificamente vastus lateralis e bíceps brachii músculos que são relativamente poupados em LGM2A (24, 25).
o tio materno era heterozigótico apenas para o CAPN3_c.550delA, e, como esperado, não apresentava quaisquer problemas neuromusculares ou cardiovasculares. O pai levou o romance CAPN3_c. 1813G > C e, assim, ele não foi afetado. Em geral, a análise da segregação confirmou a herança das três mutações do proband de seus parentes e destacou uma familiaridade para a cardiomiopatia que não pode ser negligenciada (Figura 3).
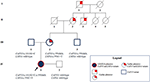
Figura 3. Pedigree mostrando a familiaridade positiva para fenótipo cardíaco herdado pela linhagem materna e a transmissão das variantes CAPN3 e LMNA em todos os membros da família.
ao todo, estes dados levantam algumas considerações importantes. Primeiro, o painel NGS neste paciente foi fundamental para alcançar o diagnóstico molecular da calpainopatia, detectando uma mutação conhecida e uma segunda variante Nova prevista como patogênica. Segundo, o painel NGS permitiu a identificação da nova variante LMNA_c.550C>T no proband. Este resultado foi consistente com a história familiar positiva e com dados de segregação, explicando doenças cardiovasculares na mãe e, mais importante, recomendando um acompanhamento cardiológico mais específico no proband. É importante notar que manifestações cardiológicas ocorreram na mãe mais tarde na idade (55 anos), de modo que podemos supor que o proband ainda poderia estar em uma “fase cardiológica assintomática”. No entanto, uma expressão fenotípica variável da mutação LMNA no proband comparada com a mãe não pode ser excluída. Todos estes dados sublinham a importância de uma abordagem integrada entre clínicos e geneticistas, para a interpretação correta dos resultados, aconselhamento genético adequado e, eventualmente, gestão clínica e acompanhamento. No que diz respeito ao aconselhamento genético e familiar, a análise da NGS também foi realizada no parceiro do proband, a fim de estimar o risco reprodutivo para o casal. O parceiro foi negativo para todos os 18 genes testados, o que significa que ele tem 1/650 risco residual para ser um portador saudável para mutações causadoras de LGMD, considerando que o teste é 84% sensível. Tendo em consideração o perfil genético do proband e a sensibilidade do teste NGS, o risco residual para o casal de ter uma criança afetada com calpainopatia é de 1/1300. Por outro lado, as variantes patogênicas LMNA são transmitidas de acordo com um padrão autossômico dominante, o que significa que o proband tem 50% de probabilidade de ter filhos heterozigóticos. No entanto, o quadro clínico da descendência não pode ser certamente previsto como o LMNA_c.550C>T é uma variante nova e seu impacto funcional no fenótipo ainda é Desconhecido.
em conclusão, este relatório de caso destaca a utilidade clínica dos painéis NGS para fornecer um diagnóstico preciso de LGMD2A e descrever fenótipos complexos e comorbidades provenientes da herança de diferentes mutações em múltiplos genes. No entanto, a aplicação da NGS, na prática clínica, deve ser sempre combinado com um pré e pós-aconselhamento genético, a fim de fornecer uma explicação clara dos resultados, as possíveis implicações em pacientes fenótipo, o risco de recorrência na família, assim como para explicar os possíveis achados inesperados.
disponibilidade de dados
não foram gerados ou analisados conjuntos de dados para este estudo.Este estudo foi realizado de acordo com as recomendações do Comité de Ética da Fundação Santa Lúcia, com o consentimento informado por escrito de todos os sujeitos. Todos os participantes deram o seu acordo por escrito, em conformidade com a Declaração de Helsínquia. O protocolo foi aprovado pelo Comitê de Ética da Fundação Santa Lúcia.
Autor Contributions
RC, CSt, VC, GC, RG, GPa, SC, CP, e JM made contributions to acquisition of data, analysis, and interpretation of data. CSt, RC, GC, RG, GM e EG estiveram envolvidos na elaboração do manuscrito. A SZ, a GPr, a CSa e a SS estiveram envolvidas na aquisição de dados clínicos. RC, CSt, VC, GC, RG, GPa, SC, CP, JM, SZ, GPr, GM, CSa, SS e EG deram a aprovação final da versão a ser publicada.
financiamento
este trabalho é apoiado pelo 5X 2016 National Health Ministry 2.
Declaração de conflito de interesses
os autores declaram que a investigação foi realizada na ausência de quaisquer relações comerciais ou financeiras que possam ser interpretadas como um potencial conflito de interesses.
material suplementar
o Material suplementar para este artigo pode ser encontrado em linha em:: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. Protein and genetic diagnosis of limb girdle muscle distrophy type 2A: the yield and the pitfalls. Nervo Muscular. (2015) 52:163–73. doi: 10.1002 / mus.24682
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Nigro V, Savarese M. Genetic basis of limb-girdle muscle distrophies: the 2014 update. Acta Myol. (2014) 33:1–12.
PubMed Abstract | Google Scholar
3. Richard I, Hogrel JY, Stockholm D, Payan CAM, Fougerousse F, Eymard B, et al. História Natural de LGMD2A para delinear as medidas de desfecho em ensaios clínicos. Ann Clin Transl Neurol. (2016) 3:248–65. doi: 10.1002 / acn3.287
PubMed Abstract / CrossRef Full Text | Google Scholar
4. Angelini C, Fanin M. Calpainopatia. In: Adam MP, Ardinger HH, Pagon RA editors. Generevews®. Seattle, WA: University of Washington (2017) 1-30.
PubMed Abstract | Google Scholar
5. Kyriakides T, Angelini C, Schaefer J, Sacconi S, Siciliano G, Vilchez JJ, et al. EFNS guidelines on the diagnostic approach to pauci – or assintomatic hyperCKemia. EUR J Neurol. (2010) 17:767–73. doi: 10.1111 / J. 1468-1331. 2010. 03012.x
CrossRef texto completo | pesquisador do Google
6. Mori-Yoshimura M, Segawa K, Minami N, Oya Y, Komaki H, Nonaka i, et al. Disfunção cardiopulmonar em doentes com distrofia muscular dos membros 2A. nervo muscular. (2017) 55:465–9. doi: 10.1002 / mus.25369
CrossRef Full Text | Google Scholar
7. Okere a, Reddy SS, Gupta s, Shinnar M. A cardiomiopatia num doente com distrofia muscular dos membros tipo 2A. (2013) 6:e12–13. D. O. I.: 10.1161 / CIRCHEARCHFAILURE.112.971424
CrossRef Full Text | Google Scholar
8. Kramerova I, Ermolova N, Eskin a, Hevener a, Quehenberger O, Armando AM, et al. Falha na regulação da transcrição dos genes necessários para a adaptação muscular subjacente à distrofia muscular 2A (calpainopatia). Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093 / hmg / ddw086
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Thompson R, Straub V. Limb girdle muscle distrophies-international collaborations for translational research. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038 / nrneurol.2016.35
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Strafella C, Caputo V, Galota MR, Zampatti S, Marella G, Mauriello S, et al. Aplicação de Medicina de precisão em doenças neurodegenerativas. Front Neurol. (2018) 9:701. doi: 10.3389 / fneur.2018.00701
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Cascella R, Strafella C, Caputo V, Errichiello V, Zampatti S, Milano F, et al. Towards the application of precision medicine in Age-Related Macular Degeneration. Prog Retin Eye Res. (2017) 63:132-46. doi: 10.1016/j. preteyeres.2017.11.004
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Cascella R, Strafella C, Longo G, Manzo L, Ragazzo M, de Felici C, et al. Avaliar o risco individual para a DMI com aconselhamento genético, história familiar e testes genéticos. Olho. (2017) 32:446–50. doi: 10.1038 / eye.2017.192
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Adams DR, Eng CM. Sequenciação da próxima geração para diagnosticar suspeitas de distúrbios genéticos. N Engl J Med. (2019) 379:1353–62. doi: 10.1056/NEJMc1814955
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Angelini C. doença Neuromuscular. Diagnóstico e descoberta em distrofia muscular. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038 / nrneurol.2015.230
CrossRef Full Text | Google Scholar
15. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett a, Reddel SW, et al. Utilização de sequenciação de Exoma completo para o diagnóstico de distrofia muscular das cintas dos membros: resultados e lições aprendidas. JAMA Neurol. (2015) 72:1424–32. doi: 10.1001 / jamaneurol.2015.2274
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Milic a, Daniele N, Lochmüller H, Mora M, Comi GP, Moggio M, et al. Um terço das biópsias LGMD2A têm uma actividade proteolítica normal de calpain 3, determinada por um ensaio in vitro. Disord De Neuromusc. (2007) 17:148–56. doi: 10.1016 / j. nmd.2006.11.001
PubMed Abstract / CrossRef Full Text | Google Scholar
17. Lanzillo R, Aurino S, Fanin M, Aguennoz m, Vitale F, Fiorillo C, et al. Calpainopatia precoce com nível normal de calpain 3 não funcional. Dev Med Child Neurol. (2006) 48:304–6. doi: 10.1017 / S001216220600065X
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Talim B, Ognibene A, Mattioli E, Richard I, Anderson LV, Merlini L. (2001) 56:692–3. doi: 10.1212 / wnl.56.5.692-a
CrossRef Full Text | Google Scholar
19. Díaz-Manera J, Llauger J, Gallardo e, Illa I. RM muscular em distrofias musculares. Acta Myol. (2015) 34:95–108.
PubMed Abstract | Google Scholar
20. Mercuri e, Bushby K, Ricci e, Birchall DR, Pane M, Kinali M, et al. Resultados de IRM muscular em doentes com distrofia muscular da cintura dos membros com deficiência em calpain 3 (LGMD2A) e contraturas precoces. Disord De Neuromusc. (2005) 15:164–71. doi: 10.1016 / j. nmd.2004.10.008
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Magri F, Nigro V, Angelini C, Mongini T, Mora M, Moroni i, et al. O registo italiano da distrofia muscular da cintura dos membros: frequência relativa, Características clínicas e diagnóstico diferencial. Nervo Muscular. (2017) 55:55–68. doi: 10.1002 / mus.25192
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Chae J, Minami N, Jin Y, Nakagawa M, Murayama K, Igarashi F, et al. Mutações genéticas de Calpain 3: resultados genéticos e clínicos patológicos na distrofia muscular dos membros. Disord De Neuromusc. (2001) 11:547–55. doi: 10.1016 / S0960-8966(01)00197-3
PubMed Abstract | CrossRef texto completo | Google Scholar
23. Richards S, Aziz N, Bale S, Bick D, Das s, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical genetic and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038 / gim.2015.30
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Bonne G, Mercuri e, Muchir a, Urtizberea a, Bécane HM, Recan D, et al. Espectro genético clínico e molecular da distrofia muscular autossómica dominante Emery-Dreifuss devido a mutações do gene lamin a/C. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170::AUXÍLIO-ANA6>3.0.CO;2-J
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
25. Hong JS, Ki CS, Kim JW, Suh YL, Kim JS, Baek KK, et al. Disritmias cardíacas, cardiomiopatia e distrofia muscular em doentes com distrofia muscular Emery-Dreifuss e distrofia muscular dos membros com cintas nos membros tipo 1B. J Sci coreana. (2005) 20:283–90. doi: 10.3346 / jkms.2005.20.2.283
CrossRef Full Text | Google Scholar