- Introdução
- Methods
- Filtragem de dados de perfis Ribossómicos
- montagem de genomas virtuais
- Determination of the Ribo-seq Read-Mapped Region on a Junction (RMRJ) of circRNAs
- formação do Modelo e classificação dos RMRJs
- Prediction of Translated Peptides by RMRJs
- Results and Discussion
- translated circRNAs in Humans and A. thaliana
- enriquecimento funcional de humanos e A. thaliana circRNAs com potencial de codificação
- Teste de Exatidão para CircCode
- Influence of the Ribo-seq Data Sequencing Depth
- Comparação de CircCode Com Outras Ferramentas
- Conclusões
- Disponibilidade e Requisitos de
- Declaração de disponibilidade de dados
- contribuições dos autores
- Financiamento
- conflito de interesses
- Material Suplementar
Introdução
Circular RNAs (circRNAs) são um tipo especial de noncoding molécula de RNA que tornou-se quente de um tema de pesquisa no campo da RNA e está recebendo uma grande atenção (Chen e Yang, 2015). Em comparação com as RNAs lineares tradicionais (contendo extremidades de 5′ e 3′), as moléculas de circRNA geralmente têm uma estrutura circular fechada, tornando-as mais estáveis e menos propensas à degradação (Vicens e Westhof, 2014). Embora a existência de cirnas tenha sido conhecida por algum tempo, estas moléculas foram consideradas como um subproduto da articulação de RNA. No entanto, com o desenvolvimento de tecnologias de sequenciamento de alta produção e bioinformática, as circRNAs tornaram-se amplamente reconhecidas em animais e plantas (Chen e Yang, 2015). Estudos recentes também mostraram que um grande número de cirnas pode ser traduzido em pequenos peptídeos nas células (Pamudurti et al., 2017) e têm papéis-chave, apesar de seu por vezes baixo nível de expressão (Hsu e Benfey, 2018; Yang et al., 2018). Embora esteja a ser identificado um número crescente de cirrnas, as suas funções em plantas e animais permanecem geralmente por estudar. Além de suas funções como Mirna decoys, circRNAs têm um potencial translacional importante, mas não existem ferramentas disponíveis para prever especificamente as capacidades translacionais dessas moléculas (Jakobi e Dieterich, 2019).
existem várias ferramentas para a previsão e identificação de circRNAs, tais como CIRI (Gao et al., 2015), CIRCexplorer (Dong et al., 2019), CircPro (Meng et al., 2017), and circtools (Jakobi et al., 2018). Entre eles, CircPro pode revelar circRNAs traduzidas calculando uma pontuação potencial de tradução para circRNAs com base em CPC (Kong et al., 2007), que é uma ferramenta para identificar o quadro de leitura aberta (ORF) em uma determinada sequência. No entanto, porque algumas circRNAs não usam o codão inicial durante a tradução (Ingolia et al., 2011; Slavoff et al., 2013; Kearse and Wilusz, 2017; Spealman et al., 2018), empregando CPC pode filtrar algumas circRNAs verdadeiramente traduzidas. Neste estudo, usamos BASiNET (Ito et al., 2018), que é um classificador de RNA baseado nos métodos de aprendizagem da máquina (floresta aleatória e modelo J48). Inicialmente transforma as RNAs de codificação (dados positivos) e as RNAs de não codificação (dados negativos) e representa-as como redes complexas; em seguida, extrai as medidas topológicas dessas redes e constrói um vetor de recurso para treinar o modelo que é usado para classificar a capacidade de codificação das circnas. Com este método, evita-se a filtragem errônea de circnas traduzidas que não são iniciadas por ago. Além disso, a tecnologia Ribo-seq, que é baseada na sequenciação de alto rendimento para monitorar RPFs (fragmentos protegidos do ribosoma) de transcrições (Guttman et al., 2013; Brar e Weissman, 2015), podem ser utilizados para determinar as localizações de cirnas que estão sendo traduzidas (Michel e Baranov, 2013). Para identificar a capacidade de codificação de circRNAs, desenvolvemos a ferramenta CircCode, que envolve um framework baseado em Python 3, e aplicamos CircCode para investigar o potencial de tradução de circRNAs de humanos e Arabidopsis thaliana. Nosso trabalho fornece um recurso rico para o estudo adicional das funções de circRNAs com capacidade de codificação.
Methods
CircCode was written in the Python 3 programming language; it uses Trimamomatic (Bolger et al.(2014), bowtie (Langmead e Salzberg, 2012) e STAR (Dobin et al., 2013) para filtrar as leituras raw Ribo-seq e mapear estas leituras filtradas para o genoma. CircCode então identifica Ribo-seq regiões mapeadas em circRNAs que contêm junções. Depois disso, as sequências mapeadas candidatas nos circRNAs são ordenadas com base em classificadores (modelo J48) em códigos RNAs e RNAs não codificados por BASiNET. Por último, os peptídeos curtos produzidos pela tradução são identificados como potenciais regiões de codificação de cirnas. Todo o processo de CircCode consiste em cinco etapas (Figura 1).
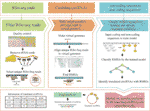
Figura 1 o fluxo de trabalho do CircCode. A camada superior representa o arquivo de entrada necessário para cada passo do CircCode. A camada média é dividida em três partes, e cada parte representa uma fase diferente de operação. Da esquerda para a direita, a primeira parte representa a filtragem dos dados Ribo-seq; o controle de qualidade é executado por Trimmatic, e as leituras rRNA são removidas por bowtie. A segunda parte representa os passos usados para produzir o genoma virtual e alinhar as leituras filtradas ao genoma virtual com a estrela. A última parte representa a identificação de circuitos traduzidos pela aprendizagem por máquina. A camada inferior representa o último passo usado para prever os peptídeos traduzidos a partir das circRNAs e os resultados finais da saída, incluindo informações sobre circRNAs traduzidas e seus produtos de tradução.
Filtragem de dados de perfis Ribossómicos
primeiro, fragmentos e Adaptadores de baixa qualidade nas leituras Ribo-Seq são removidos por Trimmomatic com os parâmetros padrão para obter leituras Ribo-seq limpas. Em segundo lugar, estas leituras Ribo-seq limpas são mapeadas para uma biblioteca rRNA para remover leituras derivadas da rRNA usando bowtie. Uma vez que os comprimentos de leitura de Ribo-seq são relativamente curtos (geralmente menos de 50 bp), é possível que uma leitura corresponda a várias regiões. Neste caso, é difícil determinar a região a que uma determinada leitura corresponde. Para evitar isso, as leituras Ribo-seq limpas são mapeadas ao genoma de uma espécie de interesse, e as leituras que não estão perfeitamente alinhadas ao genoma são consideradas como as últimas leituras Ribo-seq.
montagem de genomas virtuais
Circnas geralmente aparecem como moléculas em forma de anel em eucariotas, e elas podem ser identificadas com base em suas junções de back-splicing. No entanto, as sequências de circRNAs no arquivo fasta são muitas vezes de forma linear. Em teoria, o resultado indica que a junção está entre o nucleótido terminal de 5′ e o nucleótido terminal de 3′, embora a junção e a sequência perto da junção não possam ser vistas diretamente, assim alinhando Ribo-seq lê para sequências circRNA, incluindo junções, de uma maneira direta.
CircCode connects the sequence of each circRNA in tandem such that the junction for each is in the middle of the newly constructed sequence. Também separamos cada unidade de série por 100 N nucleótidos para evitar confusão no passo de alinhamento de sequência (o comprimento de cada RPF é inferior a 50 bp). Finalmente, obtivemos um genoma virtual que consiste apenas de circRNAs candidatas em tandem separadas por 100 Ns. Porque CircCode incide apenas sobre o alinhamento entre o Ribo-seq lê e circRNA sequências, nós podemos investigar a codificação potencial de circRNAs através do mapeamento do Ribo-seq lê para este virtual genoma, que pode salvar uma grande quantidade de tempo computacional (virtual genoma é muito menor que o genoma inteiro) e aumentar a precisão (por evitar a interferência entre a montante e a jusante comparações de sequências de circRNAs).
Determination of the Ribo-seq Read-Mapped Region on a Junction (RMRJ) of circRNAs
The final unique Ribo-seq reads read-mapped Region are mapped to a previously created virtual genome using STAR. Como cada unidade circRNA tandem foi separada por 100 bases n Antes de produzir o genoma virtual, o maior comprimento intron foi definido para não exceder 10 bases com o parâmetro “–alinhintronmax 10.”Este parâmetro elimina qualquer interação entre diferentes circRNAs no alinhamento da sequência. Na segunda etapa da produção do genoma virtual, o CircCode armazena informação de junção posicional para cada circRNA no genoma virtual. Se o Ribo-seq leia-região mapeada no virtual genoma inclui a junção de circRNA, e o número de mapeada Ribo-seq lê na junção (JNM) é maior que 3, o Ribo-seq lê-região mapeada na junção do circRNAs pode ser considerado como um RMRJ, o que revela uma traduzido aproximadamente segmento de circRNAs perto da junção do site.
formação do Modelo e classificação dos RMRJs
embora os RMRJs possam constituir uma prova poderosa de Tradução, ainda existem algumas deficiências neste método. Porque o comprimento das leituras do mapa ribossomal é curto, uma leitura pode ser comparada à posição errada. Por conseguinte, não é convincente considerar simplesmente a região abrangida pelo Ribo-seq como a região traduzida. Para este fim, o método de aprendizagem da máquina é usado para identificar a capacidade de codificação do RMRJ. Em primeiro lugar, o CircCode extrai RNAs de codificação (dados positivos) e RNAs não codificantes (dados negativos) de uma espécie de interesse e utiliza-as para a formação de modelos através da diferença nos vectores de características entre RNAs de codificação e não codificação. CircCode então usa o modelo treinado para classificar os RMRJs obtidos na etapa anterior por BASiNET. Se o RMRJ de um circRNA é reconhecido como ARN de codificação, então este circRNA pode ser identificado como um circRNA traduzido.
Prediction of Translated Peptides by RMRJs
As expression of circRNAs in organisms is low, Ribo-seq data do not show the exact 3-nt periodicity clearly in the case of fewer RPFs. Portanto, é difícil determinar o local exato de início da tradução de um circRNA traduzido. Devido à presença de um codão stop em alguns RMRJs e porque o codão é difícil de determinar, o método de encontrar uma ORF baseados em um início de codões e de um codão stop não é viável.
para determinar as verdadeiras regiões de tradução destas circRNAs e gerar o produto final de tradução, FragGeneScan (Rho et al., 2010), que pode prever regiões de codificação de proteínas em genes fragmentados e genes com andorinhas, é usado para determinar os peptídeos traduzidos produzidos por cirnas.
para evitar o processo de execução complicado, todos os modelos podem ser chamados por um script shell; o usuário pode simplesmente preencher o arquivo de configuração dado e introduzi-lo no script, e todo o processo para prever os circRNAs traduzidos será então executado. Além disso, CircCode pode ser executado separadamente, passo a passo, de modo que o usuário pode ajustar os parâmetros no meio do procedimento e ver os resultados de cada passo como desejado.
Results and Discussion
After testing on multiple computers, CircCode was found to run successfully with the required dependencies installed. Para testar o desempenho do CircCode, usamos dados para humanos e A. thaliana para prever circRNAs com potencial de tradução. Os resultados foram comparados com circnas que foram verificadas experimentalmente como confirmação. Depois disso, testamos o valor falso da taxa de descoberta (FDR) do CircCode. Usámos GenRGenS (Ponty et al., 2006) para gerar um conjunto de dados para testes com base em circRNAs traduzidas conhecidas e confirmou que o valor de FDR estava dentro de um intervalo aceitável e a um nível baixo. Finalmente, avaliamos o efeito de diferentes profundidades de sequenciamento dos dados Ribo-seq sobre as previsões de CircCode e comparamos CircCode com outros softwares.
translated circRNAs in Humans and A. thaliana
para aplicar a Ferramenta de código Circ aos dados reais, primeiro baixamos os arquivos, incluindo o genoma de referência humana GRCh38, a anotação do genoma, e rRNA humano, de Ensembl. Para A. thaliana, os genomas de referência (TAIR10), os ficheiros de anotação do genoma e as sequências rRNA correspondentes foram todos descarregados de plantas de Ensembl. Os dados Ribo-seq para os seres humanos e A. thaliana foram baixados do RPFdb (números de adesão: GSE96643, GSE81295, GSE88794) (Hsu et al., 2016; Willems et al., 2017), e todos os candidatos de human and A. thaliana foi baixada da CIRCPedia v2 (Dong et al., 2018) e a planta, respectivamente (Chu et al., 2017). Em última análise, identificamos 3,610 circRNAs traduzidas de humanos e 1.569 circRNAs traduzidas de A. thaliana usando CircCode (dados suplementares 1).
enriquecimento funcional de humanos e A. thaliana circRNAs com potencial de codificação
usando os resultados do Código Circ para humanos e A. thaliana, a ferramenta online KOBAS 3.0 (Wu et al., 2006) foi empregado para anotar estas circRNAs traduzidas com base em seus genes progenitores. Além disso, realizamos a análise funcional GO (Ontologia genética) e a análise de enriquecimento KEGG (Kyoto Encyclopedia of Genes and Genomes) para estas circRNAs traduzidas usando o pacote R clusterProfiler (Yu et al., 2012).
os resultados do KEGG mostraram que as circRNAs humanas foram enriquecidas em processamento de proteínas na Via reticulum endoplasmática, via de metabolismo do carbono e via de transporte de ARN. A análise GO indicou a participação de circRNAs humanas traduzidas na regulação da ligação das moléculas, da actividade ATPase e de outros processos biológicos relacionados com o ARN. Além disso, as circulares traduzidas de A. thaliana são enriquecidas em vias relacionadas à resistência ao estresse, sugerindo que elas desempenham papéis vitais neste processo (dados suplementares 2).
Teste de Exatidão para CircCode
Para investigar a precisão de CircCode, sequências de teste gerados pelo GenRGenS, que utiliza o modelo Markov oculto para produzir sequências que têm a mesma seqüência de características (tais como as frequências de nucleótidos diferentes, diferentes códons e nucleótidos diferentes no início da sequência), foram utilizados.
para este estudo, nós usamos circRNAs traduzidas em humanos previamente publicadas (Yang et al., 2017) como a entrada para GenRGenS e gerou 10.000 sequências para testar CircCode. Repetimos o teste 10 vezes, e, em média, 27 circRNAs traduzidas foram previstas de cada vez. O valor FDR foi calculado em 0,0027, o que é muito inferior a 0,05, indicando que os resultados previstos são credíveis.
além disso, comparamos as circRNAs traduzidas de seres humanos como identificadas pelo CircCode com dados de circRNA associados a polissomas verificados (Yang et al., 2017). Entre eles, 60% das circRNAs foram identificadas pelo Código Circ (dados suplementares 3).
Influence of the Ribo-seq Data Sequencing Depth
To investigate the impact of the sequencing depth of Ribo-seq data on the CircCode identification results, we first tested the effect of sequencing depth on the number of translated circRNAs (Figure 2A). Quando a profundidade de sequenciamento foi baixa, o número previsto de circRNAs traduzidas foi baixo, e o número de circRNAs traduzidas aumentou com o aumento da profundidade de sequenciamento. O número de circRNAs traduzidas tornou-se estável quando a profundidade de sequenciamento atingiu não menos de 10× cobertura de transcrição linear.

Figura 2 A) efeito da profundidade de sequenciamento de dados Ribo-seq sobre o número previsto de circRNAs traduzidas. B) o efeito do número de leitura da junção (JR) na sensibilidade do código de circuito a diferentes profundidades sequenciais.Em segundo lugar, foi também avaliada a influência do NMJ na sensibilidade a diferentes profundidades sequenciais (figura 2B). Os resultados mostraram que o NMJ teve menos impacto na sensibilidade à medida que a profundidade de sequenciamento aumentava. CircCode also had higher sensitivity when using Ribo-seq data with higher sequencing depth.
Comparação de CircCode Com Outras Ferramentas
Para comparar CircCode com outras ferramentas, tais como CircPro, o mesmo conjunto de Ribo-seq dados (SRR3495999) de A. thaliana foi utilizado para identificar traduzido circRNAs usando seis processadores, com 16 gigabytes de memória RAM. CircPro identificou 44 circRNAs traduzidas em 13 min, enquanto CircCode identificou 76 circRNAs traduzidas em 20 min. Assim, CircCode é mais sensível do que CircPro no mesmo nível de hardware do computador, mas leva mais tempo. CircPro é conciso e menos demorado do que CircCode, mas CircCode pode identificar mais circRNAs com capacidade de codificação do que CircPro.
Conclusões
CircRNAs desempenham um papel importante na biologia, e é crucial para identificar com precisão circRNAs com codificação capacidade para posterior investigação. Com base no Python 3, desenvolvemos o CircCode, uma ferramenta de linha de comando fácil de usar que tem alta sensibilidade para identificar circRNAs traduzidas de leitura Ribo-Seq com alta precisão. O CircCode apresenta bom desempenho em plantas e animais. O trabalho futuro adicionará a análise de caráter a jusante ao CircCode, visualizando cada etapa do processo e otimizando a precisão da previsão.
Disponibilidade e Requisitos de
CircCode está disponível em https://github.com/PSSUN/CircCode; sistema operativo(s): Linux, linguagens de programação: Python 3 e R; outros requisitos: bedtools (versão 2.20.0 ou posterior), gravata borboleta, ESTRELA, Python 3 pacotes (Biopython, Pandas, rpy2), R-pacotes (BASiNET, Biostrings). Os pacotes de instalação para todo o software necessário estão disponíveis na página inicial do CircCode. Os usuários não precisam baixá-los individualmente. A home page do CircCode também fornece manuais detalhados de usuário para referência. A ferramenta está disponível gratuitamente. Não existem restrições à utilização por não-epizootias.
Declaração de disponibilidade de dados
todos os dados relevantes estão no manuscrito e nos seus ficheiros de informação de apoio.
contribuições dos autores
conceptualização: PS, GL. Curação de dados: PS, GL. Análise Formal: PS, GL. Writing-Original Draft: PS, GL. Writing-Review and Editing: PS, GL.
Financiamento
Este trabalho foi apoiado por fundos Nacionais de Ciências Naturais da Fundação da China (grant, nº. 31770333, 31370329, e 11631012), o Programa para o Novo Século Excelentes Talentos na Universidade (NCET-12-0896), e a Investigação Fundamental Fundos para a Central de Universidades (que não. GK201403004). As agências de financiamento não tiveram qualquer papel no estudo, na sua concepção, na recolha e análise de dados, na decisão de publicar ou na preparação do manuscrito. Os financiadores não tiveram papel no desenho do estudo, na coleta e análise de dados, na decisão de publicar ou na preparação do manuscrito.
conflito de interesses
os autores declaram que a investigação foi realizada na ausência de quaisquer relações comerciais ou financeiras que possam ser interpretadas como um potencial conflito de interesses.
Material Suplementar
O Material Suplementar para este artigo podem ser encontradas online em: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
Dados Suplementares 1 | A sequência do previsto traduzido circRNA e curto peptídeo.Dados suplementares 2 / enriquecimento de GO e enriquecimento de KEGG para os seres humanos e Arabidopsis thaliana.Dados suplementares 3 / Comparação das circRNAs traduzidas previstas com circRNAs traduzidas validadas.
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: um aparador flexível para dados de sequência de iluminação. Bioinformática 30, 2114-2120. doi: 10.1093 / Bioinformática / btu170
PubMed Abstract | CrossRef Full Text | Google Scholar
Brar, G. A., Weissman, J. S. (2015). O perfil dos ribossomas revela o quê, quando, onde e como da síntese de proteínas. Conversao. Rev. Mol. Biol Celular. 16, 651–664. doi: 10.1038/nrm4069
PubMed Abstract | CrossRef Full Text | Google Scholar
Chen, L.-L., Yang, L. (2015). Regulation of circRNA biogenesis. RNA Biol. 12, 381–388. doi: 10.1080/15476286.2015.1020271
PubMed Abstract | CrossRef texto completo | Google Scholar
Chu, Q., Zhang, X., Zhu, X., Liu, C., Mao, L., Ye, C., et al. (2017). PlantcircBase: uma base de dados para RNAs circulares das plantas. Mol. Planta 10, 1126-1128. doi: 10.1016 / j. molp.2017.03.003
PubMed Abstract | CrossRef Full Text | Google Scholar
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jai, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformática 29, 15-21. doi: 10.1093 / Bioinformática / bts635
PubMed Abstract | CrossRef Full Text | Google Scholar
Dong, R., Ma, X.-K., Chen, L.-L., Yang, L. (2019). “Genome-wide annotation of circRNAs and their alternative back-splicing/splicing with CIRCexplorer Pipeline,” in Epitranscriptomics. Disfuncao. Wajapeyee, N., Gupta, R. (New York, NY: Springer New York), 137-149. doi: 10.1007/978-1-4939-8808-2_10
CrossRef Texto Completo | Google acadêmico
Dong, R., Ma, X.-K, Li, G. W., Yang, L. (2018). CIRCpedia v2: an updated database for comprehensive circular RNA annotation and expression comparison. Genómica Proteómica Bioinf. 16, 226–233. doi: 10.1016 / j. gpb.2018.08.001
CrossRef Texto Integral | Google Scholar
Gao, Y., Wang, J., Zhao, F. (2015). CIRI: um algoritmo eficiente e imparcial para identificação de RNA circular de novo. Genoma Biol. 16, 4. doi: 10.1186/s13059-014-0571-3
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Guttman, M., Russell P., Ingolia, N. T., Weissman, J. S., Lander, E. S. (2013). O perfil do ribossomo fornece evidências de que grandes RNAs não codificam proteínas. Cela 154, 240-251. doi: 10.1016/j. cell.2013.06.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Hsu, P. Y., Benfey, P. N. (2018). Pequeno mas poderoso: peptídeos funcionais codificados por pequenos ORFs em plantas. PROTEOMICS 18, 1700038. doi: 10.1002 / pmic.201700038
CrossRef Texto Completo | Google acadêmico
Hsu, P. Y., Calviello, L., Wu, H.-Y. L., Li, F. W., Rothfels, C. J., Ohler, U., et al. (2016). O perfil de super resolução de ribossomos revela eventos de tradução não anotados em Arabidopsis. Procedimento. Natl. Acad. Ciência. 113, E7126–E7135. doi: 10.1073 / pnas.1614788113
CrossRef Full Text | Google Scholar
Ingolia, N. T., Lareau, L. F., Weissman, J. S. (2011). O perfil ribossómico das células estaminais embrionárias do rato revela a complexidade e dinâmica dos proteomas dos mamíferos. Cela 147, 789-802. doi: 10.1016/j. cell.2011.10.002
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Ito, E. A., Katahira, I., Vicente, F. F., da, R., Pereira, L. F. P., Lopes, F. M. (2018). BASiNET-BiologicAl Sequences NETwork: a case study on coding and non-coding RNAs identification. Nucleic Acids Res. 46, E96-e96. doi: 10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093 / Bioinformática / bty948
CrossRef texto integral | Google Scholar
Kearse, M. G., Wilusz, J. E. (2017). Translation Non-AUG: a new start for protein synthesis in eukaryotes. Genes Dev. 31, 1717–1731. doi: 10.1101 / gad.305250.117
PubMed Abstract | CrossRef Full Text | Google Scholar
Kong, L., Zhang, Y., Ye, Z.-Q., Liu, X.-Q., Zhao, S.-Q., Wei, L., et al. (2007). CPC: avaliar o potencial de codificação de proteínas dos transcritos utilizando características de sequência e máquina de suporte vetorial. Nucleic Acids Res. 35, W345–W349. doi: 10.1093/nar/gkm391
PubMed Abstract | CrossRef Full Text | Google Scholar
Langmead, B., Salzberg, S. L. (2012). Alinhamento de leitura rápida com a Bowtie 2. Conversao. Methods 9, 357-359. doi: 10.1038 / nmeth.1923
PubMed Abstract | CrossRef Full Text | Google Scholar
Meng, X., Chen, Q., Zhang, P., Chen, M. (2017). CircPro: uma ferramenta integrada para a identificação de circRNAs com potencial de codificação de proteínas. Bioinformática 33, 3314-3316. doi: 10.1093 / Bioinformática / btx446
PubMed Abstract | CrossRef Full Text | Google Scholar
Michel, A. M., Baranov, P. V. (2013). Perfil do ribossoma: um monitor Hi-Def para síntese de proteínas na escala genoma-wide: ribosome profiling. Wiley Interdiscip. Rev. RNA 4, 473-490. doi: 10.1002 / wrna.1172
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Pamudurti, N. R., Bartok, O., Jens, M., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., et al. (2017). Tradução de Cirnas. Mol. Cela 66, 9-21.e7. doi: 10.1016 / j. molcel.2017.02.021
PubMed Abstract | CrossRef Full Text | Google Scholar
Ponty, Y., Termier, M., Denise, A. (2006). GenRGenS: software for generating random genomic sequences and structures. Bioinformática 22, 1534-1535. doi: 10.1093 / Bioinformática / btl113
PubMed Abstract | CrossRef Full Text | Google Scholar
Rho, M., Tang, H., Ye, Y. (2010). FragGeneScan: predizendo genes em leituras curtas e propensas a erros. Ácidos nucleicos Res. 38, e191-e191. doi: 10.1093/nar/gkq747
PubMed Resumo | CrossRef Texto Completo | Google acadêmico
Slavoff, S. A., Mitchell, A. J., Schwaid, A. G., Cabili, M. N., Ma, J., Levin, J. Z. et al. (2013). Descoberta peptidómica de peptídeos de leitura aberta curta codificados em células humanas. Conversao. Chem. Biol. 9, 59–64. doi: 10.1038 / nchembio.1120
PubMed Abstract | CrossRef Full Text | Google Scholar
Spealman, P., Naik, A. W., May, G. E., Kuersten, S., Freeberg, L., Murphy, R. F., et al. (2018). Uorfos não-AUG conservados revelados por uma nova análise de regressão de dados de perfil de ribossomas. Genome Res. 28, 214-222. D. N.: 10.1101 / gr.221507.117
PubMed Abstract | CrossRef Full Text | Google Scholar
Vicens, Q., Westhof, E. (2014). Biogénese de RNAs circulares. Cela 159, 13-14. doi: 10.1016/j. cell.2014.09.005
PubMed Abstract | CrossRef Full Text | Google Scholar
Willems, P., Ndah, E., Jonckheere, V., Stael, S., Sticker, A., Martens, L., et al. (2017). N-terminal proteomics assisted profiling of the unexplored translation initiation landscape in Arabidopsis thaliana. Mol. Celula. Proteomics 16, 1064-1080. doi: 10.1074 / mcp.M116. 066662
PubMed Abstract | CrossRef Full Text | Google Scholar
Wu, J., Mao, X., Cai, T., Luo, J., Wei, L. (2006). Servidor KOBAS: uma plataforma baseada na web para anotação automatizada e identificação de caminho. Nucleic Acids Res. 34, W720–W724. doi: 10.1093/nar / gkl167
PubMed Abstract / CrossRef Full Text | Google Scholar
Yang, L., Fu, J., Zhou, Y. (2018). RNAs circulares e os seus papéis emergentes na regulação imunitária. Frente. Imunol. 9, 2977. doi: 10.3389 / fimmu.2018.02977
PubMed Abstract | CrossRef Full Text | Google Scholar
Yang, Y., Fan, X., Mao, M., Song, X., Wu, P., Zhang, Y., et al. (2017). Extensive translation of circular RNAs driven by n6-methyladenosina. Cell Res. 27, 626-641. doi: 10.1038 / cr.2017.31
PubMed Abstract / CrossRef Full Text | Google Scholar
Yu, G., Wang, L.-G., Han, Y., He, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar