はじめに
非アルコール性脂肪性肝疾患(NAFLD)は、肝疾患の最も一般的な形態として認識されている(Angulo,2002;Clark et al., 2002). 非アルコール性脂肪性肝炎(NASH)は、飲酒歴がないにもかかわらず、アルコール性肝炎を模倣する(Ludwig e t a l., 1980). NAFLDおよびNASHは、肥満、インスリン抵抗性、高脂血症、および高血圧に起因するメタボリックシンドロームと関連している。 NAFLDは、最も一般的な肝疾患であると考えられ、典型的には単純な肝脂肪症として提示される(Tiniakos e t a l., 2010). 対照的に、NASHは、重度の脂肪症、小葉の炎症、および肝臓の線維症を特徴とする(Powell et al. ら,1 9 9 0;BertotおよびAdams,2 0 1 6)。 NASHの発生に関与するメカニズムは不明であるが、NASHは「最初のヒット」として肝脂肪症とその後の炎症、酸化ストレス、およびエンドトキシンなどのヒッ NASHは、メタボリックシンドロームと密接に関連しており、いくつかの臨床研究は、糖尿病、高脂血症、および高血圧の症状に焦点を当ててNASHの治療的処置を ら、2 0 0 9;Park e t a l. ら、2 0 1 0;Mahady e t a l., 2011). しかし、一般的に受け入れられている治療薬は確立されていない。
キマーゼは肝線維症の病因に関与している可能性がある。 キマーゼ活性は、線維症または肝硬変を有する患者の肝臓において有意に増加し、キマーゼレベルと線維症の程度との間に有意な相関があった(Komeda et al., 2008). キマーゼ活性の増加は、NASH患者では報告されていないが、NASHの動物モデルで観察されている(Tashiro e t a l. ら,2 0 1 0;Masubuchi e t a l. ら,2 0 1 3;Myayakaら,2 0 1 4., 2017). 対照的に、低分子阻害剤を用いたキマーゼの阻害は、ラットおよびハムスター NASHモデルにおける炎症、脂肪症および線維症の有意な減少をもたらした(Tashiro et al. ら,2 0 1 0;Masubuchi e t a l. ら,2 0 1 3;Myayakaら,2 0 1 4., 2017). これらの知見は、キマーゼがNASHの発症および進行中の炎症、脂肪症および線維症に関与している可能性があることを示している(図1)。
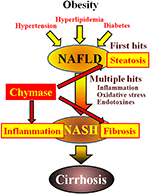
図1. NAFLDとNASHは、肥満、インスリン抵抗性、高脂血症、高血圧によってメタボリックシンドロームにリンクされています。 NASHは、肝脂肪症を「最初のヒット」とし、炎症、酸化ストレス、エンドトキシンなどの後続のヒットを伴う「複数のヒット」プロセスを介して発症すると考えられており、重度の脂肪症、炎症、線維症を特徴としています。 キマーゼは、肝臓における脂肪症、炎症、および線維症の進行に関与する可能性がある。
肥満細胞におけるキマーゼ
キマーゼのMultipul機能
キマーゼ(EC3.4.21.39)は、肥満細胞の分泌顆粒に発現する。 キマーゼは分泌か粒内で不活性なプロキマーゼとして産生され,活性化にはジペプチジルペプチダーゼi(DPPI)が必要である。 DPPIはチオールプロテイナーゼであり、最適pHは6.0である。 分泌顆粒内のpHはpH5.5で調節されるので、最適pH値はプロキマーゼを活性化するためのDPPIの提案された機能と一致している(De Young et al.,1987)(図2). しかしながら、キマーゼの最適pHは7〜9であるため、キマーゼはこのpHで肥満細胞内に酵素活性を有さない(Takai e t a l., 1996, 1997). 炎症や傷害などの刺激による肥満細胞顆粒の活性化に続いて、キマーゼが放出され、最適なpH7.4で酵素機能を発揮します(図2)。
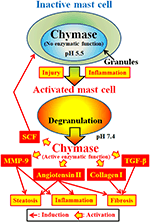
図2. キマーゼは、不活性肥満細胞の分泌顆粒に貯蔵される。 顆粒内のpHはpH5.5に維持され、キマーゼは酵素活性を有さない状態である。 キマーゼは、炎症および損傷による活性化に続いて、肥満細胞顆粒からの放出時に、アンギオテンシンII、MMP-9、TGF-β、コラーゲンIおよびSCFの形成などの酵素機能
キマーゼの複数の酵素機能
キマーゼはセリンプロテアーゼであり、一般にPhe、Tyr、Trpなどの芳香族アミノ酸の後にタンパク質のC末端側を切断する。 キマーゼは、非生物活性ペプチドアンジオテンシンiのPhe8−His9結合を切断し、ヒトを含む哺乳動物組織でその生物活性ペプチドアンジオテンシンI Iを形成することができる(Urata e t a l. ら、1 9 9 0;Takai e t a l., 1996, 1997). さらに、キマーゼは、基質メタロプロテイナーゼ(MMP)−9の前駆体を酵素的に切断し、成長因子(TGF)−βおよびコラーゲンiをそれらの活性形態に変換する(Kofford e t a l. ら、1 9 9 7;Takai e t a l. 2003年、古林ら。, 2008). さらに、キマーゼの酵素機能は、不活性な膜結合形態のSCFの酵素的切断によって幹細胞因子(SCF)を産生することができ、これは、c−kit受容体の刺激を介して未成熟, 1997). したがって、キマーゼは、アンギオテンシンII、MMP-9、TGF-β、コラーゲンI、およびSCFの活性化を含む複数の酵素機能を有する(図2)。
NASHにおけるキマーゼの酵素機能
アンギオテンシンIIは、動物NASHモデルにおけるアンギオテンシンII受容体の刺激後に活性酸素種(ROS)を増加させることによ 2007年、鍋島ら。, 2009). アンギオテンシンIIはまた、肝星細胞(Hsc)におけるα−平滑筋アクチン(SMA)の誘導を介して肝線維症を誘導した(Yoshiji et al., 2001). MMP−9は、ビトロネクチンおよびフィブロネクチンのような細胞間マトリックスの分解を介して好中球およびマクロファージの浸潤を誘導し、炎症の, 2006). NASH患者では、正常な対照と比較して肝臓でMMP-9遺伝子発現の有意な増加が観察された(Ljumovic et al., 2004). トランスジェニックマウスにおけるTGF-βの肝過剰発現は、プロコラーゲンI遺伝子発現の増強を介して重度の肝線維症を産生した(Casini et al., 1993). TGF-β形成とコラーゲンi蓄積の両方が肝線維症を誘導することが知られている。 SCFの活性化は肥満細胞数の増加を誘導し、その酵素機能は線維性組織におけるキマーゼ活性の増加をもたらす可能性がある(Maruichi e t a l., 2004). キマーゼのこれらの酵素機能は、脂肪症、炎症および線維症に関与しており、これらはすべてNASH患者および動物モデルの肝臓で観察される(図2)。
NASH動物モデルにおけるキマーゼの関与
メチオニンおよびコリン欠損(MCD)食は、典型的なNASHモデルを誘導するために広く使用されている。 Mcd食餌を与えたハムスターでは、血漿中の総ビリルビン、トリグリセリド、およびヒアルロン酸の有意な増加が観察された(Tashiro et al., 2010). さらに,肝臓では炎症細胞の蓄積と脂質沈着面積と線維性面積の増加が観察された。 このMCD食誘導NASHモデルでは、肝キマーゼ活性およびアンギオテンシンII、MMP−9およびコラーゲンiのような関連因子が有意に増加した(Tashiro e t a l. ら,2 0 1 0;Masubuchi e t a l., 2013). 最近、脳卒中を起こしやすい自然発症高血圧5/Dmcr(SHRSP5/Dmcr)ラットに高脂肪およびコレステロール(HFC)食を与えた新しいNASHモデルが開発されました(Kitamori et al., 2012). このモデルは、NASH患者の症状に臨床的に類似していると考えられるメタボリックシンドロームの症状を示した(Kitamori et al., 2012). HFC食誘導NASHモデルでは,高血圧および高脂血症が観察され,肝臓に重度の脂肪症,線維症および炎症性細胞蓄積が検出された(Miyaoka et al., 2017). さらに、肝におけるmmp-9、TGF-β、およびコラーゲンIと共にキマーゼ活性の有意な増強が観察された(Miyaoka et al., 2017). したがって、NASHの動物モデルにおけるキマーゼとNASH病因との間には密接な関係があるようである。
NASH動物モデルにおけるキマーゼ阻害剤の効果
NASH動物モデルにおけるキマーゼ阻害剤の効果
低分子キマーゼ阻害剤は、mcd飼料NASHハムスターモデルでは、mcd食と同時に阻害剤の投与を開始したときに、キマーゼ活性を有意に減衰させ、肝臓におけるアンギオテンシンII、MMP-9およびコラーゲンIレベルを減少させた(Tashiro et al. ら,2 0 1 0;Masubuchi e t a l., 2013). キマーゼ阻害剤は、このNASHモデルにおける肝脂肪症、線維症、および炎症性細胞の蓄積を有意に防止した(Tashiro et al. ら,2 0 1 0;Masubuchi e t a l., 2013). 酸化ストレスはNASH発生の”多重ヒット”理論において役割を果たすと考えられ,酸化ストレスマーカーのマロンジアルデヒドの増強はキマーゼ阻害剤によって肝臓で有意に弱毒化された(Masubuchi et al., 2013). ハムスター MCD食誘導NASHモデルにおいて、キマーゼ阻害剤は、確立されたNASH中で投与されたときに改善効果を示した(Masubuchi e t a l., 2013). 肝臓における脂肪症および線維症の両方の程度は、キマーゼ阻害剤の投与前と比較して減少した(Masubuchi et al., 2013).
高血圧ラットHFC食誘導NASHモデルの肝臓では、低分子キマーゼ阻害剤は、すべてキマーゼ関連因子であるキマーゼならびにMMP-9、TGF-βおよびコラーゲンIのレベ, 2017). キマーゼ阻害剤は、肝脂肪症および線維症を有意に減衰させ、炎症、特に好中球浸潤のマーカーとしてのミエロペルオキシダーゼを減少させた(Miyaoka et al., 2017). このHFC食誘発モデルでは、プラセボ投与群の生存率は、HFC食の開始後1 4週間で0%であり、重度の肝不全に起因した(Miyaka e t a l., 2017). しかし、hfc食の開始直後にキマーゼ阻害剤でラットを処理したキマーゼ阻害剤処理群は、100週で14%の生存を示した。 さらに、キマーゼ阻害剤で処理したラットについて、HFC食餌摂食の開始から8週間後に5 0%の生存率が報告され、その時点でNASHが確立された(Myaka e t a l., 2017).
したがって、キマーゼ阻害剤は、動物モデルにおけるNASHの予防および改善のための有用な薬剤であり得る。 一方、アンギオテンシンIIはまた、間接的にMMP-9およびTGF-β遺伝子発現の増加を介して肝炎症、脂肪症、および線維症を促進する。 MMP−9およびTGF−βは共に、NASHの病因に密接に関与しているが、これらの因子は、必ずしもアンギオテンシンIIのみによって誘導されるわけではない(Takai e t a l., 2010). アンギオテンシンII刺激以外の因子は、MMP−9およびTGF−β遺伝子発現の増加に寄与する(Takai e t a l., 2010). このような場合、アンギオテンシンII受容体遮断薬(ARB)はMMP-9およびTGF-β作用を減衰させることができないが、キマーゼ阻害剤はMMP-9およびTGF-β活性化の阻害を介して減衰効果を有する可能性があり、NASH進行の予防のための潜在的な治療経過を示す。
キマーゼ阻害剤によって弱毒化された肝炎症のメカニズム
キマーゼ阻害剤は、ハムスター MCD食およびラットHFC食誘導NASHモデルにおける炎症を軽減するこ ら,2 0 1 0;Masubuchi e t a l. ら,2 0 1 3;Myayakaら,2 0 1 4., 2017). キマーゼ阻害剤の治療は、肝臓におけるキマーゼ活性を有意に減衰させ、アンギオテンシンIIおよびMMP-9レベルを減少させた(Tashiro et al. ら,2 0 1 0;Masubuchi e t a l. ら,2 0 1 3;Myayakaら,2 0 1 4., 2017). HSCでは、アンギオテンシンIIは、ニコチンアミドアデニンジヌクレオチドリン酸(NADPH)オキシダーゼの活性化を介して過酸化水素やスーパーオキシドなどのROS生成を誘導する(De Minicis And Brenner、2007)。 キマーゼ阻害剤は、ハムスター MCD誘導NASHモデルにおけるアンギオテンシンIIレベルの減少に加えて、NADPHオキシダーゼ成分Rac-1および酸化ストレスマーカーマロンジア, 2013). ROSのアンジオテンシンI I誘導増強は、好中球およびマクロファージにおけるMMP−9遺伝子発現を促進した(Yaghoti e t a l. ら,2 0 1 1;Kurihara e t a l., 2012). したがって、キマーゼ阻害剤は、直接MMP-9にproMMP-9の活性化を阻害し、間接的に減少したアンギオテンシンIIを介してMMP-9遺伝子発現を減少させます。 MMP−9は、ビトロネクチンおよびフィブロネクチンのような細胞外マトリックス成分を切断し、肝臓の完全性の崩壊を導き、マクロファージおよび好中球の浸潤を誘導する(Medina e t a l., 2006). HFC食誘導NASHモデルでは,マクロファージおよび好中球におけるミエロペルオキシダーゼ発現の有意な増加が肝臓で観察され,キマーゼ阻害剤によって減少した(Miyaoka et al., 2017). したがって、キマーゼ阻害剤によって弱毒化炎症のメカニズムは、肝臓におけるアンギオテンシンIIおよびMMP-9レベルの減少に依存し得る。
キマーゼ阻害剤によって弱まる肝脂肪症のメカニズム
アンギオテンシンIIは、ROS産生を介して肝脂肪症に影響を与える可能性があります。 マウスH SCでは、NADPHオキシダーゼ阻害剤がROS産生を有意に減少させ、ARBは、ros産生の減少を介して肝脂肪症の発症を遅らせた(Hirose e t a l. ら、2 0 0 7;Guimaranes e t a l., 2010). MCD食誘導NASHマウスモデルでは、アンジオテンシンII受容体欠損マウスにおいて脂肪症の有意な減衰が観察された(Nabeshima e t a l., 2009). インビボおよびインビトロ実験の両方で、アンギオテンシンIIは、ステロール調節要素結合タンパク質(SREBP)-1cおよび脂肪酸シンターゼ(FAS)遺伝子発現をアップレギュレー ら,2 0 0 1;Hongo e t a l., 2009). 対照的に、ARBは、マウスNASHモデルにおけるROS減衰を介してsrebp−1cおよびFASの遺伝子発現をダウンレギュレートするとともに、肝脂肪症を弱毒化した(Kato e t a l., 2012). ハムスター MCD食誘導NASHモデルにおいて、SREBP−1cおよびFAS遺伝子発現の有意な減衰が、低分子キマーゼ阻害剤による処理後に観察された(Masubuchi e t a l., 2013). したがって,キマーゼ阻害剤による肝脂肪症の改善機序は,肝臓におけるアンギオテンシンII生成の減少によるROS産生の減少に依存すると考えられる。
キマーゼ阻害剤によって減衰される肝線維症のメカニズム
キマーゼは、非生物活性前駆体TGF-βからのTGF-βの形成に寄与し、TGF-βは線維芽細胞の増殖を強く誘導することが知られていることから、組織線維症の進行と密接に関連している可能性がある(Takai et al. ら,2 0 0 3;Oyamada e t a l., 2011). TGF−βは、活性化HSCを介してNASH患者における線維症の進行において中心的な役割を果たすことが知られている(Williams e t a l., 2000). 遺伝子発現およびシグナル伝達を介したTGF−β機能の阻害は、実験モデルにおいて改善された肝線維症をもたらした(George e t a l. ら、1 9 9 9;Ariasら、1 9 9 9;Ariasら、, 2003). ラットHFC食誘導NASHモデルにおいて、キマーゼ阻害剤によるキマーゼ活性の減衰は、肝臓におけるTGF-βレベルおよび線維化領域の減少をもたらした(Miyaoka et al., 2017). 従って,キマーゼ阻害剤によるTGF-βの減少は肝線維症の予防に寄与する可能性がある。
アンギオテンシンIIは肝線維症の誘導にも関与している可能性がある。 アンジオテンシンIIは、HSCの収縮および増殖を誘導し、また、in vitroで線維芽細胞におけるTGF−βの遺伝子発現を誘導する(Kagami e t a l. ら、1 9 9 4;Bataller e t a l., 2000). 野生型マウスでは胆管結さつによってTGF-βレベルとコラーゲン蓄積および線維性病変の程度の両方が観察されたが、これらはアンギオテンシンII受容体欠損マウスでは弱毒化された(Yang et al., 2005). ラットNASHモデルでは、ARBはまた、TGF-β遺伝子発現の減少を介して肝線維症を弱毒化した(Hirose et al., 2007). アンギオテンシンI I誘導TGF-β遺伝子発現以外のアンギオテンシンI Iと肝線維症との間にも関係がある可能性がある。 慢性C型肝炎患者では、ARBは、Rac−1遺伝子発現を介してコラーゲン遺伝子発現を減少させた(Colmenero e t a l., 2009). HSCは肝臓におけるコラーゲンの主要産生細胞として認識されており,hscにおけるα-平滑筋アクチン(SMA)の発現の増強はコラーゲンiを含む細胞外マトリックス沈着を強く誘導する(De Minicis And Brenner,2007)。 アンジオテンシンIIはラットHSCにおいてα-SMA遺伝子発現を誘導することができる。 対照的に、アンギオテンシンII遮断は、α−SMAの減少と共に肝線維症の減少をもたらす(Yoshiji e t a l., 2001). NASH患者では評価されなかったが,肝硬変患者の肝臓の線維化領域ではキマーゼとアンギオテンシンI I形成活性の両方が有意に増強され,キマーゼ,アンギオテンシンI I形成活性と肝線維化の間に有意な相関が認められた(Komeda e t a l., 2008). ハムスター四塩化物誘発性肝硬変モデルでは、キマーゼおよびアンギオテンシンII形成活性の有意な増加が観察され、これは低分子キマーゼ阻害剤による処, 2010).
肥満細胞安定剤トラニラストは、肥満細胞の活性化を阻害し、キマーゼの放出を遮断し、それによってラット糖尿病およびHFC食誘導NASHモデルにおける肝線維症の発症を防止することができる(Uno et al., 2008). キマーゼは、その酵素機能によるSCF活性化を介して肥満細胞の増殖を促進する(Longley e t a l., 1997). NASH動物モデルでは、キマーゼ阻害剤は肝臓における肥満細胞数の増加を減少させ、キマーゼ阻害剤による直接阻害後のキマーゼ活性の低下および肥満細胞 ら,2 0 1 3;Myayakaら,2 0 1 4., 2017).
したがって、キマーゼ阻害剤は、チマーゼ阻害によるTGF-β活性化の阻害および/またはアンギオテンシンIIおよび肥満細胞増殖の減少を介したTGF-βレベルの減
おわりに
肥満、インスリン抵抗性、高脂血症、高血圧からなるメタボリックシンドロームは、NASHの開発と密接に関連しており、NASHの治療のために抗糖尿病、抗高脂血症、抗高血圧薬の試験が行われている。 これらの薬剤の背後にある概念は、代謝症候群の症状を減弱させることである(Georgescu e t a l. ら、2 0 0 9;Park e t a l. ら、2 0 1 0;Mahady e t a l., 2011). 以前の報告は、キマーゼ阻害剤が、それぞれ、糖尿病、高脂血症、および高血圧の動物モデルにおいて、血糖および脂質レベルおよび血圧に影響を及ぼすこ ら、2 0 0 9;Takai e t a l. ら、2 0 1 4;Zhang e t a l., 2016). したがって、キマーゼ阻害の背後にある概念は、nashの肝炎症および線維症を直接減衰させることである。 我々は、メタボリックシンドロームを標的とするキマーゼ阻害剤は、NASHの進行の減衰のための潜在的に強力な戦略であることを提案します。
著者の貢献
STとDJ:原稿を書いた。 両方の著者は最終的な原稿を読み、承認した。
利益相反に関する声明
著者らは、本研究は、利益相反の可能性があると解釈される可能性のある商業的または財政的関係がない場合に行われた