- Introduction
- CHREBP構造とLID/Graceドメインを介した調節
- グルコース代謝産物によるChREBPの活性化
- ChREBP Co-factor and Partners
- 炭水化物代謝およびヘパトカイン産生におけるChREBPの役割
- 肝脂肪酸合成およびVLDL分泌の調節因子としてのChREBP
- 肝臓および腸におけるChREBPによるフルクトース代謝の調節
- BDKの規制:肝臓におけるPPM1K軸
- ChREBPは、Fgf21のグルコースを介した調節に必要とされる
- エネルギー恒常性を制御する臓器間ネットワークにおけるChREBPの役割
- インスリン感受性バランスの制御における肝ChREBPの役割
- 脂肪ChREBPは、インスリン感受性に脂肪形成をリンク
- 脂肪組織におけるホルモン感受性リパーゼとChREBPの新規相互作用
- 結論と今後の方向性
- 著者の貢献
- 資金調達
- 利益相反声明
Introduction
近年、ショ糖や高果糖コーンシロップなどの単糖の消費が増加し、肥満、脂質異常症、2型糖尿病および/または非アルコール性脂肪肝疾患(NAFLD)などの代謝性疾患のリスクが増加している。 肝臓は、過剰な食物炭水化物の脂肪への変換を担う主要な器官である。 生じるトリグリセリド(TG)は非常に低密度のリポ蛋白質(VLDL)に詰まり、循環に分泌するか、脂質のしぶきとして貯えられるか、またはベータ酸化の細道を通 血糖値の上昇に応答して分泌されるインスリンは、転写因子ステロール調節要素結合タンパク質-1c(SREBP-1c)を介してde novo脂肪酸合成(lipogenesis)の遺伝子の発現を刺激する(Foretz et al., 1999). SREBP-1cは、炭水化物応答要素結合タンパク質(ChREBP)と呼ばれる別の転写因子との相乗効果で作用し、これは食物炭水化物に対する応答を仲介する。 Chrebpタンパク質構造は、低グルコース阻害ドメイン(LID)と、そのN末端に位置するグルコース応答活性化保存要素(GRACE)を含む(Li e t a l., 2006). グルコース代謝産物によるGRACEドメインの活性化は、ChREBP転写活性を促進し、炭水化物応答要素(雑用)と呼ばれる高度に保存された配列への結合を促進する。 L−ピルビン酸キナーゼ(L−pk)、解糖における律速酵素、脂肪酸合成酵素(Fa S)、アセチル−Coaカルボキシラーゼ(Acc)およびステアロイル−Coaデサチュラーゼ(Scd1)(Kawakughe e t a l.,2 0 0 2)を含むdenovo lipogenesisのキー酵素をコードするChrebp標的遺伝子のプロモーター上に雑用が存在する(Kawakughe e t a l.,2 0 0 2)。, 2001). 最近の研究では、肝臓における解糖および脂肪原性遺伝子発現の完全な誘導のためのChREBP(グルコースによって活性化)とSREBP-1c(インスリンによって活性化)との間の相互依存性が報告されている(Linden et al., 2018). Chrebp欠損マウス(ChREBPKO)の肝臓におけるsrebp-1cの核活性型のウイルス再確立は、解糖遺伝子発現を救出に影響を与えないが、脂肪原性遺伝子発現を正規化した。 Srebp-1cノックアウトマウスの肝臓でChREBP発現が誘導されたミラー実験は、脂肪酸合成遺伝子の制御におけるChREBPのよく知られている役割にもかかわらず、解 それにもかかわらず、この研究は、脂肪酸合成の調節に関与する対照遺伝子におけるChREBPおよびSREBP-1cの二重作用の重要性を示唆している(Linden et al., 2018).
ChREBPは肝臓で高度に濃縮されており、脂質代謝のマスターレギュレータとして研究されている(Iizuka et al. ら,2 0 0 4;Osorio e t a l., 2016). また、CHREBPは、膵島、小腸、骨格筋においても有意に発現され、腎臓および脳においてはより少ない程度で発現される(Richards e t a l. 2017年、レビューのために)。 興味深いことに、ChREBPの別のアイソフォーム、Chrebp Βは、代替の第一エクソンプロモーターに由来し、脂肪組織で最初に同定された(Herman et al. 他の細胞型に記載されている(Abdul−Wahed e t a l.,2 0 1 2を参照されたい)。 2017年、レビューのために)。 Chrebp Βは構成的に活性なアイソフォームとして記述される。 今後の研究は、グルコースおよび脂質代謝の調節におけるChREBPおよびChrebp Βのそれぞれの役割に対処するだけでなく、それらの特定および/または重複するター
CHREBP構造とLID/Graceドメインを介した調節
ChREBPはbHLH/Zip転写因子のMondoファミリーに属しています。 N末端ドメイン(1-251残基)は、二つの核輸出シグナル(NES)ともexportin1および/または14-3-3タンパク質と呼ばれる染色体領域維持1(CRM1)と相互作用することによ, 2008). C末端領域は、共因子およびDNA結合に関連するポリプロリンドメイン、BHLH/LZドメイン(6 6 0〜7 3 7残基)およびロイシンジッパー様ドメイン(ジップ様、8 0 7〜8 4 7残基)を含 ら,2 0 0 1;Fukasawa e t a l. ら、2 0 1 0;Ge e t a l., 2012). ChREBPの局在化および転写活性化は、栄養素の利用可能性によって決定される。 ChREBPのグルコースを介した調節は、導入(図1A)で述べたように、lidとGRACEドメインで構成されているグルコースセンシングモジュール(GSM)またはmondo保存領域(MCR)のレベ; Li et al. ら、2 0 0 6年;SinghおよびIrwin、2 0 1 6年)。 2012年、Herman et al. (2012)は、代替の第一エクソンプロモーター1bからエクソン2に転写される別のChREBPアイソフォーム、Chrebp Βを記載した(図1B)。 この転写産物はエクソン4から翻訳され、687アミノ酸の短いタンパク質を生成する(全長ChREBPアイソフォームはαと改名され、原稿ではChREBPと呼ばれる864アミノ酸を含む)。 CHREBP βは、GLUT−4依存的に白色脂肪組織中で高度に活性であり、雑用配列がエキソンプロモーター1βで同定されたので、Chrebpaによって直接調節されることが示唆されている(Herman e t a l.,2012;図1B). ChREBPaによるChrebp Βの調節は可能性としてはhyperglycemic条件の下でブドウ糖への応答を悪化させる供給前方ループの存在を提案する。 しかし、Chrebp Βアイソフォームの調節機構、およびより重要なのは、その特定の機能を解明する必要があります。
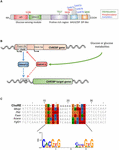
図1. (A)炭水化物応答要素結合蛋白質α(Chrebpa)の構造。 ChREBPaは864のアミノ酸で構成され、複数の規定する範囲を含んでいます。 N末端で蛋白質は低いブドウ糖の抑制的な範囲(LID)およびブドウ糖によって活動化させる保存された要素(GRACE)で構成されるブドウ糖感知モジュールを含 このタンパク質はまた、ポリプロリンリッチ、bHLH/LZおよびc末端に位置するロイシンジッパー様(ジップ様)ドメインを含む。 翻訳後修飾は、それぞれの残基、リン酸化(赤)、アセチル化(青)および最近同定されたO-GlcNAcylations(緑)に示されている。 (B)ChREBP遺伝子の遺伝子構造および二つのChREBPアイソフォームαおよびβの生成。 この転写産物は、エクソン4から翻訳され、二つのNES、NLSおよびLIDドメインが欠落している687アミノ酸の短いタンパク質を生成する。 Chrebp Βアイソフォームは、エクソンプロモーター1bで雑用配列が同定されたため、ChREBPaによって直接調節されることが示唆されている。ChREBP αおよびβアイソフォームの両方が雑用に結合するかどうかは現在知られていない。 Herman e t a l. (2012). (C)雑用コンセンサス配列のマルチアライメントは、いくつかのChREBP標的遺伝子プロモーターに提示します。 ヌクレオチドに基づくアラインメントは、Poungvarin et al. (2015). この特定の整列に関連するコンセンサス配列に対応するロゴもまた表される。
グルコース代謝産物によるChREBPの活性化
絶食条件下では、プロテインキナーゼA(PKA)のグルカゴン依存的活性化(Kawaguchi et al. ら、2 0 0 2)残基Ser1 9 6およびThr6 6 6上のChrebpをリン酸化し、タンパク質1 4−3−3へのChrebp結合および細胞質ゾル中のその保持をもたらす(Kawakughe e t a l. ら,2 0 0 1,2 0 0 2;Davies e t a l., 2008). AMP活性化プロテインキナーゼ(AMPK)、中央細胞エネルギーセンサーはまた、残基Ser5 6 8上のChrebpをリン酸化し、これは、次に、その標的遺伝子のプロモーターへのChrebpの結合を減少させる(Kawakughe e t a l. ら,2 0 0 2;Satoら,2 0 0 2;Satoら,, 2016). 脂肪酸酸化から産生されるAMPおよびケトン体などの絶食中に生成される代謝産物は、ChREBPおよび14-3-3タンパク質親和性を変化させ、複合体の安定化を 2008年、中川ら。 ら,2 0 1 3;Satoら,2 0 1 3;Satoら,, 2016). 炭水化物に応答して、ChREBPは、転写、翻訳および翻訳後のレベルで調節される。 食事後のグルコース濃度の増加は、最初にプロテインホスファターゼ2A(PP2A)の活性化剤として提案されたキシルロース-5-リン酸(X5P)などの中間代謝産物の合成を促進する(Kawaguchi et al. ら,2 0 0 1;Kabashima e t a l., 2003). PP2Aは、以前に、それがX5P−およびPP2A−依存的に(Thr6 6 6およびSer5 6 8上で)さらに脱リン酸化される核へのその転座を可能にするSer1 9 6残基でChrebpを脱リン しかし、このモデルは長年にわたって挑戦され、グルコース-6-リン酸(G6P)などの他の代謝産物は、ChREBP転座/活性の潜在的な活性化剤であることが提案された(Dentin et al., 2012). McFerrin et al. ら(2 0 1 2)は、Chrebp/MondobオルソログであるMondoaでも保存されているGRESSドメイン上のG6P結合(2 5 3−SDTLFT−2 5 8)の推定モチーフを同定した(Richards e t a l. 2017年、レビューのために)。 この仮説によれば、G6Pは、Chrebpの開放立体配座を誘導するアロステリック立体配座の変化を促進し、補因子との相互作用およびその後の核への転座を促進することができる(Mcferrin e t a l., 2012).
核内では、ChREBPはグルコース代謝に依存する翻訳後修飾であるO-GlcNAcylationを介して修飾することができ、ChREBP転写活性に重要であることが同定されている(Guinez et al., 2010). O-GlcNAcylationは、o-GlcNAcトランスフェラーゼ(OGT)、それによってその活性、安定性および/または細胞内の位置を変更するタンパク質をターゲットにN-アセチルグルコサミン(GlcNAc)残基 Yangら。 (2017)は最近、O-GlcNacylationによって修飾されたいくつかのChREBP残基を明らかにした。 BHLH/ZIPおよび二量体化および細胞質位置ドメイン(DCD)ドメインにおけるこれらの残基の変異は、Chrebpのグルコース依存性活性化のための必須部位としてThr517お ChREBPはまた、p3 0 0のヒストンアセチルトランスフェラーゼ活性を介したアセチル化によって修飾することもできる(Bricambert e t a l., 2010). グルコース活性化p300は、Lys672上のChREBPをアセチル化し、最適なコンセンサス結合配列であるCaygycnnnnncrcrtgである雑用配列へのリクルートを強化することにより、その転写活性を増加させる(図1C)。 Poungvarin et al. (2015)は、高炭水化物、無脂肪食を再給餌したマウスの肝臓および白色脂肪組織におけるChIP-seqによるChREBP結合部位を分析した。 彼らは、chrebp結合がインスリンシグナル伝達、接着接合および癌に関与する経路に富むことを報告し、腫瘍形成および癌進行におけるChREBPの新規な関与を示唆している。 さらに、最近の研究では、肝細胞癌(HCC)におけるChrebpの重要性が報告されている(Ribback e t a l., 2017). その結果、ChREBPの遺伝的欠失(ChREBPKOマウス)が、マウスのプロテインキナーゼB/Akt過剰発現によって誘発される肝発癌を障害することが分かった。 さらに、マウスおよび/またはヒトHCC細胞におけるChREBPのsiRNA媒介阻害は、増殖およびアポトーシスの減少をもたらした。
ChREBP Co-factor and Partners
Chrebpのいくつかのco-factor and/or partnersが過去数年間にわたって同定された(Richards et al. 2017年、レビューのために)。 Max like protein x(Mlx)、BHLH/LZ転写因子は、Mondoファミリーの共通の結合パートナーとして最初に同定された(Stoeckman e t a l., 2004). MlxとChREBPの二量体化は、グルコースに応答して核転座と雑用要素への結合の両方に必要です。 肝細胞核因子4α(Hnf4A)およびファルネソイドX受容体(FXR)などの核受容体もChREBPパートナーとして記載されていた。 HNF4aは、Chrebp標的遺伝子のプロモーター上の直接反復1(DR−1)領域に結合することによって、Chrebpと物理的に相互作用する(Adamson e t a l. ら、2 0 0 6;Meng e t a l., 2016). さらに、p3 0 0/CBP転写共活性化因子タンパク質は、Chrebp/Hnf4A複合体を安定化させることが示された(Burke e t a l., 2009). P300/CBP転写共活性化タンパク質は、転写装置と複数のシグナル依存性イベントを調整し、統合する上で中心的な役割を果たしています。 P300/CBPのもう一つの主特性はヌクレオソームのヒストンの調整によってクロマチンの活動に影響を与える容量のp300/CBPに寄与するヒストンのアセチルトランスフェラーゼ(HAT)の活動の存在である。 ヒト肝細胞では、ChREBP-Hnf4A複合体に結合するFXRは、CBP/p300からのChREBPの放出を誘発し、lpkプロモーター上のヒストン脱アセチラーゼSMRTの募集につながり、それによってChREBP転写活性の共リプレッサーとして作用する(Caron et al., 2013). 加えて、CBP/p3 0 0H AT活性は、Lys6 7 2上のChrebpを改変し、グルコースに応答してその転写活性化をもたらす(Bricambert e t a l., 2010).
Bricambert et al. (2018)は最近、chrebpの新規補因子として、ヒストンリジンデメチラーゼ(KDM7)ファミリーに属するヒストンデメチラーゼ植物homeodomain finger2(Phf2)を同定した。 Phf2とChREBPの間の相互作用は、その標的遺伝子のプロモーター上のH3K9メチルマークを消去することによってChREBP転写活性化を強化します。 興味深いことに、Phf2およびChrebpの、核性赤血球2様2因子(Nrf2)のプロモーターへの特異的な同時動員は、高血糖の状況における活性酸素種(ROS)およびNAFLD進行の増, 2018).
炭水化物代謝およびヘパトカイン産生におけるChREBPの役割
肝脂肪酸合成およびVLDL分泌の調節因子としてのChREBP
非アルコール性脂肪肝疾患はメタボリックシンドロームの特徴であり、ヒトにおける研究では、de novo lipogenesisがNAFLD患者の全肝脂質の約25%に寄与していることが明らかになっている(Donnelly et al., 2005). インスリン抵抗性状態では、高血糖および高インスリン血症は、ChREBPおよびSREBP-1cの活性化によって部分的に脂肪形成を増強する。 肥満およびインスリン抵抗性ob/obマウスの肝臓におけるCHREBP阻害は、Rnaiまたは遺伝的アブレーションを介して、肝脂肪症の逆転をもたらす(Dentin e t a l. 2006年、飯塚ら。, 2006). 肝臓によるVLDLの分泌の変化もまた、NAFLDの病因に寄与する。 ミクロソームトリグリセリド移動蛋白質(MTTP)は,アポリポタンパク質B含有リポ蛋白質の組み立てと分泌を担当する蛋白質である。 マウスおよびヒトにおけるMTTPの欠乏は、低脂質血症および脂肪肝を引き起こす。 このタンパク質の調節は、重要な正および負の調節ドメインを含む、そのプロモーター中のいくつかの高度に保存されたシス要素と関連している(Cuchel et al. ら、2 0 1 3;Hussain e t a l., 2011). 最近、肝臓における機能的Chrebpの欠如がMttp発現およびVLDL組立および分泌を抑制することから、ChrebpはMTTPの潜在的な調節因子として指摘された(Niwa et al., 2018). しかし,Mttpプロモーター上では雑用が明確に同定できなかったため,ChrebpがMttpを調節する機構を同定するためにはさらなる解析が必要である。
肝臓および腸におけるChREBPによるフルクトース代謝の調節
ChREBPとフルクトース代謝の間のリンクは、ChREBPノックアウトマウス(ChREBPKOマウス)の表現型解析によ ChrebPKOマウスは、高フルクトース食(Hfrd)摂食の数日以内に死亡することが報告された(Iiuzaka e t a l., 2004). フルクトースに対するこの主要な不耐性は、フルクトース代謝に必要な二つの酵素であるフルクトキナーゼおよびトリオースキナーゼの発現の減少に起因す, 2004). Kim et al. (2016)は後に、肝臓および全身フルクトースクリアランスにおけるフルクトースのグルコースへの効率的な変換のためのChREBPの重要性を報告したが、また、フルクトースの摂取下で、ChREBPは直接トランス活性化g6Pc発現、糖新生の重要な遺伝子によって高血糖に寄与する可能性がある。 この効果は、フルクトースの消費がChrebp活性であるがグルコース産生を悪化させる悪循環につながる可能性がある(Kim e t a l., 2016). 翌年、張らによる研究。 (2017)は、HFrDを与えられたChREBPKOマウスは、小胞体ストレスとCCAATエンハンサー結合タンパク質相同タンパク質(CHOP)媒介肝細胞アポトーシスの過剰活性化に起因する重 これらのマウスの肝細胞におけるアポトーシスは、hmg-CoAレダクターゼ(HMGCR)またはSREBP2阻害を介してこの経路の阻害は、HFrD誘発性肝損傷からChREBPKOマウスを救出したので、最も可能性の高いコレステロール生合成の増加にリンクされていた。 ChREBPの欠如はまた、最近、マウスにおける糖不耐性および吸収不良につながるショ糖およびフルクトース代謝の調節不全に関連していた(Kato et al., 2018). これらの効果は、グルコースとフルクトース中のスクロースを消化する腸ショ糖イソマルターゼ(SI)、グルコース輸送体5(Glut5)と2(Glut2)、およびフルクト分解を調節するケトヘキナーゼ(Khk)酵素の発現低下と関連していた(図2)。 これらの酵素の調節不全は、消化されていないショ糖およびフルクトースの蓄積につながり、腸内微生物叢組成に潜在的な影響を及ぼす可能性がある。 Chrebpkoとhfrdを与えられた肝臓特異的ChREBPノックアウト(ChREBPLiverKO)マウスとの比較は、以前に肝ChREBP欠乏症だけではフルクトース不耐性につながることはありませんが、小腸でChREBP欠, 2017). 全体として、これらの研究は、フルクトース代謝の調節におけるChREBPの重要性を強調し、小腸におけるその役割と調節のより良い理解の必要性を強調する。
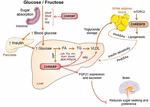
図2. ChREBPは、グルコースおよびフルクトースに応答して複数のシグナル伝達/代謝経路を調節する。 ChREBPは腸、レバーおよび白い脂肪組織を含む複数のティッシュで表現されます。 これらの細胞型では、グルコースおよび/またはフルクトースに応答して、ChREBPが活性化され、図に示すように特定のgenicプログラムを誘導する。 腸では、CHREBPによるSI、Glut5、Glut2およびケトヘキソキナーゼ(Khk)発現の刺激(直接的または間接的に)は、ショ糖耐性およびフルクトース吸収を改善するために記載さ 肝臓では、ChREBPは、それによって肝臓からの脂肪酸蓄積とVLDLの輸出の両方を制御し、解糖、脂肪生成およびミクロソームトリグリセリド転送タンパク質(Mttp) ChREBPはまた調整します繊維芽細胞の成長因子21(FGF21)のようなhepatokinesの生産をあります。 この肝臓から脳への軸は、肝臓における基質の取り扱いだけでなく、栄養選好にも影響を与える肝調節因子から全身調節因子に肝臓ChREBP機能を拡張する。 白色脂肪組織におけるChREBP活性化は、保護循環信号を生成することによって代謝恒常性の改善にリンクされています。 脂肪酸とヒドロキシ脂肪酸(パルミチン酸ヒドロキシルステアリン酸)との間の分岐エステル結合によって特徴付けられるほ乳類脂質の新しいクラスは、β細胞機能の直接およびインクレチンを介した変調、脂肪グルコース取り込みを強化し、炎症を減少させることにより、グルコース恒常性に有益な効果を発揮することが報告された。 興味深いことに、mtorc2は最近、脂肪細胞におけるChrebp Βアイソフォームの新規レギュレータとして同定された。
BDKの規制:肝臓におけるPPM1K軸
分岐鎖アミノ酸(BCAAs)異化の最初のコミットされたステップは、分岐鎖α-ケト酸デヒドロゲナーゼキナーゼ(BDK)とプロテインホスファターゼ、Mg2+/Mn22+依存1K(PPM1K)によって制御される分岐鎖ケト酸デヒドロゲナーゼ(BCKDH)複合体によって調節される。 ホワイト他 (2018)最近、ChREBPは肝臓におけるBDKのアップレギュレーションおよびPPM1Kのダウンレギュレーションと関連し、これらの遺伝子の両方のプロモーターに保存された雑用モチーフを同定した。 高グルコースまたはフルクトース食餌を与えたラットの肝臓において,BDKの発現と他の典型的なChrebp標的遺伝子(Fasn,Pklr,Chrebp Β)との間に正の相関が観察された。 生理学的なレベルでは、BDKの増加:PPM1Kの比率はそれによりde novoのlipogenesisを刺激するATPクエン酸塩のリアーゼ(ACLY)のリン酸化そして活発化をもたらしました。 これらの知見は、BDKとPPM1KはChrebp Βによって調節される新規な脂肪形成活性化遺伝子である可能性があることを明らかにした。 脂質、グルコースおよびアミノ酸代謝の調節におけるそれらの役割を考えると、BDKおよびPPM1Kは、近い将来、肝臓における潜在的な治療標的と考えられ, 2018).
ChREBPは、Fgf21のグルコースを介した調節に必要とされる
ChREBPは、最近、線維芽細胞成長因子21(FGF21)などのヘパトカインの産生および分泌に関連していた(Iizuka et al. ら、2 0 0 9;Dushay e t a l. ら、2015、Iroz et al., 2017). FGF2 1は、肝臓によって合成された代謝ホルモンであり、末梢組織において複数の有益な効果を有する(Kharitonenkov e t a l. ら、2 0 0 5;Badman e t a l. ら、2 0 0 7;Markan e t a l., 2014). 最近まで、fgf21は、ペルオキシソーム増殖剤活性化受容体α(PPARa)の転写制御下で脂肪酸酸化、ケトン生成および脂肪分解を促進する絶食ホルモンと考えられていた(Inagaki et al., 2007). Fgf2 1プロモーターの雑用は、マウス(−7 4〜−5 2bp)およびヒト(−3 8 0〜−3 6 6bp)の両方において以前に同定されている(Iiuzaka e t a l.,2009)しかし、機能的研究は最近まで不足しています。 グルコースおよびフルクトースの消費は、健康なボランティアおよびメタボリックシンドローム患者においてFGF2 1レベルの急速な上昇をもたらすことが, 2015). 追加の研究はまた、肝臓−脳軸を介したChrebp由来のFGF2 1と多量栄養素の選好との間の機構的な関連も報告した(Talukdar e t a l. 2016年;-フォン-ホルスタイン-Rathlou et al., 2016). この肝臓から脳への軸は、肝代謝調節因子から全身調節因子にChREBP機能を拡張し、肝臓基質の取り扱いだけでなく、全体的な摂食行動にも影響を与える(Abdul-Wahed et al., 2017).
エネルギー恒常性を制御する臓器間ネットワークにおけるChREBPの役割
インスリン感受性バランスの制御における肝ChREBPの役割
私たちの研究室では、ChREBPが非アルコール性およびアルコール性肝疾患の文脈における肝脂肪酸組成およびインスリン感受性の重要なモジュレーターとして作用することが以前に報告されている(Abdul-Wahed et al. 2017年、レビューのために)。 ChREBPを過剰発現マウスは、コントロールよりも大きな肝脂肪症を開発したが、興味深いことに代謝合併症の自由に滞在し、インスリン抵抗性を開発し 脂質分析により、Crebp媒介性脂肪症は、飽和脂肪酸の減少および一価不飽和脂肪酸の増加と関連し、後者は、インスリン感受性に対するCrebp媒介性有益な効果, 2012). これらの結果は、脂質分配におけるChrebpの役割を実証し、特定の脂質種が、適切な位置および時間に存在する場合、代謝ストレスへの適応を調節するシ ら、2 0 1 2;Bricambert e t a l., 2018). 興味深いことに、Jois e t a l. (2017)はまた、全身グルコース恒常性およびインスリン感受性に関する肝ChREBPの保護的役割を示唆した。 肝脂肪症から保護しながらChREBPLiverKOマウスは、悪化した耐糖能を示します。 肝Crebp欠失はまた,白色および褐色脂肪組織における遺伝子発現変化をもたらし,組織間コミュニケーションを示唆した。 従って、全身エネルギーバランスへのChrebpの寄与は、エネルギー恒常性の組織間協調に寄与する脂質種および/または肝細胞産生のその調節に依存し得る(Jois e t a l., 2017).
脂肪ChREBPは、インスリン感受性に脂肪形成をリンク
脂肪組織におけるインスリンシグナル伝達の障害は、インスリン抵抗性の重要な特徴である。 研究は、白色脂肪組織におけるChrebp活性化が、保護循環シグナルを産生することによって代謝恒常性を改善することができることを報告している(Yore et al. ら、2 0 1 4;Tang e t a l., 2016). 脂肪酸とヒドロキシ脂肪酸、パルミチン酸ヒドロキシルステアリン酸(PAHSA)との間の分岐エステル結合によって特徴付けられる哺乳動物脂質のクラスは、β細胞機能、グルコース取り込みおよび炎症の減少の直接およびインクレチン媒介変調を介してグルコース恒常性に有益な効果を発揮することが報告された(Yore et al., 2014). 同様に、脂肪組織の低いlipogenesis率を表わす脂肪質特定のChREBPのノックアウト(ChREBPadiposeKO)はchowおよび高脂肪の食事療法の条件の両方の下でレバー、筋肉および白い脂肪組織の損なわれたインシュリンの行為とインシュリン抵抗力があるインシュリン抵抗力があります。 ChrebPadiposekoマウスは、血清中のPahsasレベルが低い一方で、PAHSA補充、特に9−PAHSA異性体は、chrebpadiposeko全体的なインスリン抵抗性および脂肪組織炎症を救済し、脂肪−Chrebpの損失がインスリン抵抗性を引き起こすのに十分であることを確認している(Vijayakumar e t a l., 2017). 最近の研究では、脂肪細胞におけるChREBP(特にβアイソフォーム)の新規レギュレータとしてラパマイシン複合体2(mtorc2)の機構標的を同定した。 成熟脂肪細胞におけるrapamycin-insensitive companion of mTOR(Rictor)の特定のアブレーションは、Chrebp Βのダウンレギュレーションおよび脂肪形成制御に関与する標的遺伝子発現につながる脂肪組織, 2016). ChREBPによって媒介される重要な脂肪–肝臓クロストークと一致して、これらの効果は、肝臓のインスリン抵抗性と強化された糖新生と関連している。 全体として、これらの研究は、インスリン感受性シグナルの誘発における脂肪Chrebpの重要な役割を支持する(Tang e t a l., 2016).
脂肪組織におけるホルモン感受性リパーゼとChREBPの新規相互作用
ChREBPは最近、脂肪組織における脂肪分解酵素ホルモン感受性リパーゼ(HSL)のパートナーとし, 2019). ヒト脂肪細胞およびマウス脂肪組織におけるHSLのノックダウンは、インスリン感受性を高め、非常に長鎖脂肪酸酵素(Elovl6)の伸長を誘導することが示 Elov1 6は、C1 2−1 6飽和および一価不飽和脂肪酸の伸長をChrebp依存的に調節するミクロソーム酵素である(Morigny e t a l., 2019). 機構レベルでは、HSLとChrebpとの間の物理的相互作用は、Chrebpaの核転座、およびその後のChrebp Βおよび標的遺伝子、特にElovl6の誘導を障害した(Morigny e t a l., 2019). 本研究では、脂肪組織におけるChREBPのための新規な調節を明らかにする。 HSLとChREBP間の相互作用を阻害することは、脂肪細胞におけるインスリン感受性を改善するための潜在的な治療戦略につながる可能性があります。
結論と今後の方向性
ChREBPは現在、確立された炭水化物センサーです。 ほとんどの調査が解糖およびlipogenic細道の制御の含意に専用されていたが、最近のデータはまたhepatokinesおよび/またはlipokinesの作成で器械であることができるhepatocytesと脂肪細胞のChREBPの新しい貢献を明らかにした臓器間の混線を誘発する。 議論されたように、これらの組織における新たに同定された補助因子(エピジェネティック修飾因子)および/またはパートナー(脂肪HSL)はまた、NAFLDおよび/また 最近の研究はまた、フルクトース代謝の調節におけるChREBPの重要性を支持し、小腸におけるその役割と調節のより良い理解の必要性を強調している。 最後に、重要な細胞型におけるChREBPaおよびChrebp Βの特異的および/または重複する標的を特定するだけでなく、インスリン感受性に対する特異的な影響を決定することは、今後数年間で特に重要になるであろう。
著者の貢献
リストされているすべての著者は、仕事に実質的な、直接的かつ知的な貢献をし、出版のためにそれを承認しました。
資金調達
Posticのラボ(U1016-Institut Cochin)は、ChroMEネットワーク(Marie Curie Skłodowska Action H2020-MSCA-ITN-2015-675610)、医学研究財団(FRM)(DEQ20150331744)、ANR-15-CE14-0026-
利益相反声明
著者らは、この研究は潜在的な利益相反と解釈される可能性のある商業的または財政的関係がない場合に行われたと宣言している。