はじめに
環状Rna(circrna)は、RNAの分野でホットな研究テーマとなり、大きな注目を集めている非コードRNA分子の特殊なタイプです(Chen and Yang,2015)。 従来の線状Rna(5’末端および3’末端を含む)と比較して、circRNA分子は通常、閉じた円形構造を有し、それらをより安定にし、分解しにくい(Vicens and Westhof、2014)。 Circrnaの存在はしばらくの間知られていましたが、これらの分子はRNAスプライシングの副産物であると考えられていました。 しかし、ハイスループットシーケンシングとバイオインフォマティクス技術の発展に伴い、circrnaは動物や植物で広く認識されるようになりました(Chen and Yang、2015)。 最近の研究では、多数のcircRNAが細胞中の小さなペプチドに翻訳され得ることも示されている(Pamudurti e t a l. る(Hsu and Benfey,2 0 1 8;Yang e t a l.,2 0 1 7)。, 2018). 増加する数のcircRNAが同定されているが、植物および動物におけるそれらの機能は一般的に研究されるべきである。 MiRNAデコイとしての機能に加えて、circrnaは重要な翻訳可能性を持っていますが、これらの分子の翻訳能力を具体的に予測するためのツールはありません(Jakobi and Dieterich、2019)。
ciriのようなcircrnaの予測と同定のためのいくつかのツールが存在する(Gao et al. ら、2 0 1 5)、Circexplorer(Dong e t a l. ら、2 0 1 9)、Circpro(Meng e t a l. ら、2 0 1 7)、およびcirctools(Jakobi e t a l., 2018). その中でも、CircProは、CPCに基づいてcircRNAの翻訳電位スコアを計算することによって、翻訳されたcircRNAを明らかにすることができる(Kong et al. Orf(open reading frame)を特定するためのツールである。 しかし、一部のcircRNAは翻訳中に開始コドンを使用しないため(Ingolia e t a l. ら、2 0 1 1;Slavoff e t a l. ら,2 0 1 3;KearseおよびWilusz,2 0 1 7;Spealmanら.,2018),CPCを採用すると、いくつかの真の翻訳circrnaを除外することができます. 本研究では、BASiNET(Ito et al. これは、機械学習法(random forestおよびJ48モデル)に基づくRNA分類器である。 最初に与えられたコーディングRna(正のデータ)と非コーディングRna(負のデータ)を変換し、それらを複雑なネットワークとして表し、その後、これらのネットワークのトポロジカル測度を抽出し、circrnaのコーディング能力を分類するために使用されるモデルを訓練するための特徴ベクトルを構築する。 この方法では、AUGによって開始されない翻訳されたcircRNAの誤ったフィルタリングが回避される。 さらに、転写物のRpf(リボソームで保護された断片)をモニターするための高スループット配列決定に基づくRibo−seq技術(Guttman e t a l. ら、2 0 1 3;Brar and Weissman,2 0 1 5)を利用して、翻訳されているcircRNAの位置を決定することができる(Michel and Baranov,2 0 1 3)。 Circrnaのコーディング能力を同定するために、我々はPython3ベースのフレームワークを含むツールCircCodeを開発し、ヒトとシロイヌナズナからcircrnaの翻訳可能性を調査するためにCircCodeを適用した。 私たちの仕事は、コーディング能力を持つcircrnaの機能のさらなる研究のための豊富なリソースを提供します。
メソッド
CircCodeはPython3プログラミング言語で書かれており、Trimmomaticを使用しています(Bolger et al. ら、2 0 1 4)、bowtie(LangmeadおよびSalzberg、2 0 1 2)、およびSTAR(Dobin e t a l. 生のRibo-seq読み取りをフィルタリングし、これらのフィルタリングされた読み取りをゲノムにマッ その後、CircCodeは、ジャンクションを含むcircrnaのRibo-seq読み取りマップされた領域を識別します。 その後、circRNA中の候補マッピング配列は、分類器(J4 8モデル)に基づいて、BasinetによってコードRnaおよび非コードRnaに分類される。 最後に、翻訳によって生成された短いペプチドは、circrnaの潜在的なコード領域として同定される。 CircCodeのプロセス全体は5つのステップで構成されています(図1)。
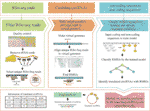
図1CircCodeのワークフロー 最上位層は、CircCodeの各ステップに必要な入力ファイルを表します。 中間層は3つの部分に分かれており、各部分は異なる操作段階を表しています。 左から右へ、最初の部分はRibo-seqデータのろ過を表します;品質管理はTrimmomaticによって実行され、rRNAはbowtieによって読まれます取除かれます。 第二の部分は、仮想ゲノムを生成し、フィルタリングされた読み取りを仮想ゲノムにSTARで整列させるために使用されるステップを表す。 最後の部分は、機械学習による翻訳されたcircrnaの同定を表しています。 一番下の層は、circRNAから翻訳されたペプチドと、翻訳されたcircRNAとその翻訳産物に関する情報を含む最終出力結果を予測するために使用される最後のス
リボソームプロファイリングデータのフィルタリング
最初に、Ribo-Seq読み取りの低品質のフラグメントとアダプタをtrimmomaticによってデフォルトのパラメータで削除して、クリーンなRibo-seq読み取りを取得します。 第二に、これらのきれいなRibo-seq読み取りは、bowtieを使用してrRNAから派生した読み取りを除去するためにrRNAライブラリにマッピングされます。 Ribo-seqの読み取り長さは比較的短い(一般に50bp未満)ため、1つの読み取りが複数の領域に一致する可能性があります。 この場合、特定の読み出しがどの領域に対応するかを決定することは困難である。 これを避けるために、きれいなRibo-seq読み取りは、関心のある種のゲノムにマッピングされ、ゲノムに完全に整列していない読み取りは、最終的なユニークなRibo-seq読み取りとみなされます。
仮想ゲノムの組み立て
Circrnaは通常、真核生物ではリング状の分子として現れ、それらのバックスプライシング接合に基づいて同定することができる。 しかしながら、FASTAファイル中のcircRNAの配列は、多くの場合、線状の形態である。 理論的には、この結果は、接合部が5’末端ヌクレオチドと3’末端ヌクレオチドの間にあることを示しているが、接合部と接合部付近の配列は直接見ることができないため、Ribo-seqは接合部を含むcircRNA配列に簡単に整列させることができる。
CircCodeは、各circRNAの配列をタンデムで接続し、それぞれの接合部が新しく構築された配列の中央にあるようにします。 また、配列アライメントステップでの混乱を避けるために、各シリーズ単位を100Nヌクレオチドで分離しました(各RPFの長さは50bp未満です)。 最後に、我々は100Nsによって分離されたタンデムで候補circrnaのみからなる仮想ゲノムを得た。 CircCodeはRibo-seq読み取りとcircRNA配列のアライメントのみに焦点を当てているため、Ribo-seq読み取りをこの仮想ゲノムにマッピングすることにより、circrnaのコーディング可能性を調べることができ、大量の計算時間を節約し(仮想ゲノムは全ゲノムよりもはるかに小さい)、精度を向上させることができる(circrnaの上流と下流の配列比較の間の干渉を回避することによって)。
circrnaの接合部(RMRJ)上のRibo-seq Read-Mapped領域の決定
最終的なユニークなRibo-seq読み取りは、STARを使用して以前に作成された仮想ゲノムにマッピングされます。 各タンデムcircRNAユニットは、仮想ゲノムを生成する前に100N塩基によって分離されたので、最大イントロン長は、パラメータ”–alignIntronMax10で10塩基を超えないように設”このパラメータは、配列アライメントにおける異なるcircrna間の相互作用を排除します。 仮想ゲノム生成の第二段階では、CircCodeは仮想ゲノム内の各circRNAの位置接合情報を格納する。 仮想ゲノム中のRibo-seq read-mapped領域がcircRNAの接合部を含み、マッピングされたRibo-seq reads on junction(NMJ)の数が3より大きい場合、circrnaの接合部上のRibo-seq reads-mapped領域はRMRJとみなすことができ、接合部付近のcircrnaの大まかに翻訳されたセグメントを明らかにする。
RMRJsのモデルと分類のトレーニング
RMRJsは強力な翻訳証明を構成することができますが、この方法にはまだいくつかの欠点があります。 リボソームマップの読み取りの長さが短いため、読み取りは間違った位置と比較される可能性があります。 したがって、単にRibo-seq読み取りによってカバーされる領域を翻訳された領域と考えることは説得力がありません。 この目的のために、機械学習方法は、RMRJの符号化能力を識別するために使用される。 まず、CircCodeは、対象種からコードRna(陽性データ)と非コードRna(陰性データ)を抽出し、コードRnaと非コードRnaの特徴ベクトルの違いによってモデルトレーニングに使用します。 次に、CircCodeは学習済みモデルを使用して、前の手順でBASiNETによって取得されたRMRJsを分類します。 CircRNAのRMRJがコードRNAとして認識される場合、このcircRNAは、翻訳されたcircRNAとして同定することができる。
RMRJsによる翻訳されたペプチドの予測
生物におけるcircrnaの発現が低いため、Ribo-seqデータはRpfが少ない場合に正確な3nt周期性を明確に示さない。 したがって、翻訳されたcircRNAの正確な翻訳開始部位を決定することは困難である。 いくつかのRmrjには停止コドンが存在し、開始コドンを決定することが困難であるため、開始コドンおよび停止コドンに基づいてORFを見つける方法は実<4 0 3 5><3 5 6>これらのcircRNAの真の翻訳領域を決定し、最終翻訳産物を生成するために、Fraggenescan(Rho e t a l. ら、2010)は、断片化された遺伝子およびフレームシフトを有する遺伝子のタンパク質コード領域を予測することができ、circrnaによって産生される翻訳されたペプ
面倒な実行プロセスを避けるために、すべてのモデルをシェルスクリプトで呼び出すことができます。 さらに、CircCodeは、ユーザーが手順の途中でパラメータを調整し、必要に応じて各ステップの結果を表示できるように、段階的に別々に実行することができます。
結果と考察
複数のコンピュータでテストしたところ、必要な依存関係がインストールされているCircCodeが正常に実行されていることがわかりました。 CircCodeの性能をテストするために、ヒトとa.thalianaのデータを使用して、翻訳可能性のあるCircRNAを予測しました。 結果を確認として実験的に検証されたcircRNAと比較した。 その後、CircCodeの偽発見率(FDR)値をさらにテストしました。 本発明者らは、Genrgens(Ponty e t a l. 既知の翻訳されたcircRNAに基づく試験のためのデータセットを生成し、FDR値が許容可能な範囲内でかつ低レベルであることを確認した。 最後に、CircCode予測に対するRibo-seqデータの異なる配列深度の効果を評価し、CircCodeを他のソフトウェアと比較しました。
ヒトおよびAにおける翻訳されたcircrna。 thaliana
CircCodeツールを実際のデータに適用するために、まずEnsemblからヒト参照ゲノムGrch38、ゲノムアノテーション、ヒトrRNAを含むファイルをダウンロードしました。 A.thalianaの場合、参照ゲノム(TAIR10)、ゲノムアノテーションファイル、および対応するrRNA配列はすべてEnsembl植物からダウンロードされました。 ヒトおよびa.thalianaのRibo−seqデータを、Rpfdb(受託番号:GSE9 6 6 4 3、GSE8 1 2 9 5、GSE8 8 7 9 4)からダウンロードした(Hsu e t a l. ら、2016;Willemsら、2016。 ら、2 0 1 7)、およびヒトおよびA由来のすべての候補circRNAを含む。 thalianaは、Circpedia v2(Dong e t a l. ら、2 0 1 8)およびPlantcircbase(Chu e t a l., 2017). 最終的に、我々はCircCode(補足データ1)を使用して、ヒトから3,610翻訳circrnaとA.thalianaから1,569翻訳circrnaを同定した。
ヒトおよびA.thalianaのCircrnaのコード化可能性を有する機能的濃縮
ヒトおよびA.thalianaのCircCode結果を用いたオンラインツールKOBAS3.0(Wu et al. ら,2 0 0 6)を用いて、それらの親遺伝子に基づいてこれらの翻訳されたcircRNAに注釈を付けた。 さらに、R package clusterProfilerを用いて、これらの翻訳されたcircRNAのGO(Gene Ontology)機能解析とKEGG(Kyoto Encyclopedia of Genes and Genomes)エンリッチメント解析を行いました(Yu et al., 2012).
keggの結果は、ヒトcircrnaが小胞体経路、炭素代謝経路、およびRNA輸送経路におけるタンパク質処理に富むことを示した。 GO分析は、分子結合、ATPase活性、および他のRNAスプライシング関連の生物学的プロセスの調節におけるヒト翻訳circrnaの関与を示した。 さらに、a.thalianaの翻訳されたcircrnaは、ストレス耐性に関連する経路に富み、このプロセスにおいて重要な役割を果たしていることを示唆している(補足データ2)。
Circcodeの精度テスト
CircCodeの精度を調べるために、genrgensによって生成されたテストシーケンスは、隠されたマルコフモデルを使用して同じ配列特性(異なるヌクレオチド、異なるコドン、シーケンスの開始時の異なるヌクレオチドの周波数など)を持つ配列を生成するために使用された。
本研究では、以前に公開されたヒト翻訳circrnaを使用しました(Yang et al. 2017)をGenRGenSの入力として生成し、CircCodeをテストするために10,000個のシーケンスを生成しました。 我々は、試験を1 0回繰り返し、平均して、2 7回の翻訳されたcircRNAが毎回予測された。 FDR値は0.0027と計算され、これは0.05よりもはるかに小さく、予測結果が信頼できることを示しています。
さらに、CircCodeによって同定されたヒト由来の翻訳されたcircRNAと、検証されたポリソーム関連circRNAデータとを比較した(Yang et al., 2017). その中で、circRNAの60%がCircCodeによって同定された(補足データ3)。
Ribo-seqデータ配列決定深度の影響
Ribo-seqデータの配列決定深度がCircCode同定結果に及ぼす影響を調べるために、最初に翻訳されたcircrnaの数に及ぼす配列決定深度の影響を試験した(図2A)。 配列決定の深さが低いとき,翻訳されたcircRNAの予測数は低く,翻訳されたcircRNAの数は配列決定の深さの増加とともに増加した。 翻訳されたcircrnaの数は、配列決定の深さが10×線状転写カバレッジ以上に達したときに安定になった。

図2(A)翻訳されたcircRNAの予測数に対するRibo−seqデータ配列決定深度の効果。 (B)異なるシーケンス深さでのCircCode感度に対するジャンクション読み取り番号(JRN)の影響。
第二に、異なる配列決定深さでの感度に対するNMJの影響も評価した(図2B)。 結果は、配列決定の深さが増加するにつれて、NMJが感度に及ぼす影響が少ないことを示した。 CircCodeはまた、より高い配列深度を有するRibo-seqデータを使用すると、より高い感度を有していた。
CircCodeと他のツールとの比較
CircCodeをCircProなどの他のツールと比較するために、A.thalianaの同じRibo-seqデータ(SRR3495999)を使用して、16ギガバイトのRAMを持つ六つのプロセッサを使用して翻訳されたcircrnaを同定した。 CircProは44分で翻訳されたcircrnaを13分で同定したが、CircCodeは76分で翻訳されたcircrnaを20分で同定した。 したがって、CircCodeは同じコンピュータハードウェアレベルでCircProよりも敏感ですが、より多くの時間がかかります。 CircProはCircCodeよりも簡潔で時間がかかりませんが、CircCodeはCircProよりもコーディング能力を持つより多くのcircrnaを識別することができます。
結論
Circrnaは生物学において重要な役割を果たしており、その後の研究のためにコード能力を持つcircrnaを正確に同定することが重要である。 Python3をベースに、Ribo-Seq読み取りから翻訳されたcircrnaを高精度に識別するための高感度な使いやすいコマンドラインツールCircCodeを開発しました。 CircCodeは植物および動物両方の良い業績を表わす。 今後の作業では、プロセスの各ステップを視覚化し、予測の精度を最適化することにより、下流の文字分析をCircCodeに追加します。
可用性と要件
CircCodeはhttps://github.com/PSSUN/CircCodeで利用可能です;オペレーティングシステム:Linux,プログラミング言語:Python3およびR;その他の要件:bedtools(バージョン2.20.0以降),bowtie,STAR,Python3パッケージ(Biopython,Pandas,rpy2),R-packages(BASiNET,Biostrings)。 必要なすべてのソフトウェアのインストールパッケージは、CircCodeのホームページで入手できます。 ユーザーは個別にダウンロードする必要はありません。 CircCodeのホームページには、参照用の詳細なユーザーマニュアルも用意されています。 このツールは自由に利用できます。 非アカデミックによる使用に制限はありません。
データ可用性ステートメント
関連するすべてのデータは、原稿とそのサポート情報ファイル内にあります。
著者の貢献
概念化:PS、GL。 データキュレーション:PS、GL。 形式的な分析:PS、GL。 執筆-元の草案:PS、GL。 執筆-レビューと編集:PS、GL。
資金調達
本研究は、中国国家自然科学財団(助成番号31770333、31370329、および11631012)、新世紀大学優秀人材育成プログラム(NCET-12-0896)、中央大学基礎研究資金(no. GK201403004)。 資金調達機関は、研究、その設計、データ収集と分析、出版の決定、または原稿の準備には役割を果たしていませんでした。 資金提供者は、研究デザイン、データ収集と分析、出版の決定、原稿の準備には何の役割もありませんでした。
利益相反
著者らは、この研究は潜在的な利益相反と解釈される可能性のある商業的または財政的関係がない場合に行われたと宣言しています。
補足資料
この記事の補足資料はオンラインで見つけることができます: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
補足データ1/予測された翻訳されたcircRNAおよび短いペプチドの配列。
補足データ2|ヒトおよびシロイヌナズナのGO濃縮およびKEGG濃縮結果。
補足データ3|予測された翻訳されたcircrnaと検証された翻訳されたcircrnaの比較。
Bolger,A.M.,Lohse,M.,Usadel,B.(2014). Trimmomatic:illuminaのシーケンスデータのための適用範囲が広いトリマー。 バイオインフォマティクス30,2114-2120. doi:10.1093/バイオインフォマティクス/btu170
PubMed要約/CrossRef全文/Google Scholar
ることができます。 リボソームプロファイリングは、タンパク質合成の何を、いつ、どこで、どのように明らかにする。 ナット モル牧師 セルバイオール 16, 651–664. doi:10.1038/nrm4069
PubMed要約/CrossRef全文/Google Scholar
Chen,L.-L.,Yang,L.(2015). CircRNAの生物発生の調節。 RNA Biol. 12, 381–388. 土井: 10.1080/15476286.2015.1020271
PubMed Abstract|CrossRef全文|Google Scholar
Chu,Q.,Zhang,X.,Zhu,X.,Liu,C.,Mao,L.,Ye,C.,et al. (2017). PlantcircBase:植物の環状Rnaのデータベース。 モル 植物10、1126-1128。 土井:10.1016/j.molp.2017.03.003
PubMed要約/CrossRef全文/Google Scholar
Dobin,A.,Davis,C.A.,Schlesinger,F.,Drenkow,J.,Zaleski,C.,Jha,S.,et al. (2013). スター:超高速ユニバーサルRNA-seqアライナー。 バイオインフォマティクス29,15-21. doi:10.1093/バイオインフォマティクス/bts635
PubMed要約/CrossRef全文/Google Scholar
Dong,R.,Ma,X.-K.,Chen,L.-L.,Yang,L.(2019). “Genome-wide annotation of circRNAs and their alternative back-splicing/splicing with CIRCexplorer Pipeline,”In Epitranscriptomics. Eds. Wajapeyee,N.,Gupta,R.(ニューヨーク、NY:Springer New York),137-149. 土井: 10.1007/978-1-4939-8808-2_10
CrossRef全文|Google Scholar
Dong,R.,Ma,X.-K.,Li,G.-W.,Yang,L.(2018). CIRCpedia v2:包括的な環状RNA注釈と発現比較のための更新されたデータベース。 ゲノミクスプロテオミクスバイオインフ。 16, 226–233. doi:10.1016/j.gpb.2018.08.001
CrossRef全文|Google Scholar
Gao,Y.,Wang,J.,Zhao,F.(2015). チリ: de novo環状RNA同定のための効率的かつ公平なアルゴリズム。 ゲノムバイオール 16, 4. ドイ:10.1186/s13059-014-0571-3
PubMed Abstract|CrossRef全文|Google Scholar
Guttman,M.,Russell,P.,Ingolia,N.T.,Weissman,J.S.,Lander,E.S.(2013). リボソームプロファイリングは、大きな非コードRnaがタンパク質をコードしないという証拠を提供する。 セル154、240-251。 土井:10.1016/j。2013.06.009
PubMed要約/CrossRef全文/Google Scholar
ることができます。 小さいながらも強大な: 植物の小さいOrfによってコードされる機能ペプチッド。 プロテオミクス18、1700038。 ドイ:10.1002/pmic.201700038
CrossRef全文|Google Scholar
Hsu,P.Y.,Calviello,L.,Wu,H.-Y.L.,Li,F.-W.,Rothfels,C.J.,Ohler,U.,et al. (2016). 超解像リボソームプロファイリングは、シロイヌナズナのunannotated翻訳イベントを明らかにする。 プロク… ナトル アカド サイ… 113,E7126-E7135. ドイ:10.1073/pnas.1614788113
CrossRef全文/Google Scholar
インゴリア、N.T.、Lareau、L.F.、Weissman、J.S.(2011年)。 マウス胚性幹細胞のリボソームプロファイリングは、哺乳類のプロテオームの複雑さとダイナミクスを明らかにする。 郵便番号は147-789-802。 土井:10.1016/j。2011.10.002
PubMed要約/CrossRef全文/Google Scholar
2018年、伊藤,E.A.,片平,I.,Vicente,F.F.,da,R.,Pereira,L.F.P.,Lopes,F.M.(2018). BASiNET-BiologicAl Sequences NETwork:a case study on coding and non-coding Rna identification. Nucleic Acids Res.46,e96–e96. 土井:10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093/バイオインフォマティクス/bty948
CrossRef全文/Google Scholar
ることができます。 非AUG翻訳:真核生物におけるタンパク質合成のための新しいスタート。 遺伝子Dev. 31, 1717–1731. 土井:10.1101/gad.305250.117
Pubmed要約/CrossRef全文/Google Scholar
Kong,L.,Zhang,Y.,Ye,Z.-Q.,Liu,X.-Q.,Zhao,S.-Q.,Wei,l.,et al. (2007). CPC:配列機能とサポートベクターマシンを使用して転写物のタンパク質コードの可能性を評価します。 核酸Res.35,W345-W349. 土井:10.1093/nar/gkm391
PubMed要約/CrossRef全文/Google Scholar
Langmead,B.,Salzberg,S.L.(2012). Bowtie2との速いギャップ読まれた直線。 ナット 方法9、357-359。 ドイ:10.1038/nmeth.1923
PubMed Abstract/CrossRef Full Text|Google Scholar
Meng、X.、Chen、Q.、Zhang、P.、Chen、M.(2017)。 CircPro:蛋白質コードの潜在性のcircrnaの同一証明のための統合された用具。 バイオインフォマティクス33,3314-3316. 土井:10.1093/バイオインフォマティクス/btx446
PubMed要約/CrossRef全文|Google Scholar
Michel,A.M.,Baranov,P.V.(2013). リボソームプロファイリング:aこんにちは-Defモニターのためのタンパク質合成における全ゲノム規模でのリボソームプロファイリング. ワイリー-インターディシップ 第4巻、473-490頁。 ドイ:10.1002/wrna.1172
PubMed要約/CrossRef全文/Google Scholar
Pamudurti,N.R.,Bartok,O.,Jens,M.,Ashwal-Fluss,R.,Stottmeister,C.,Ruhe,L.,et al. (2017). CircRNAsの翻訳。 モル セル66、9-21。e7 土井:10.1016/j.molcel.2017.02.021
PubMed要約/CrossRef全文/Google Scholar
Ponty,Y.,Termier,M.,Denise,A.(2006). GenRGenS:ランダムなゲノム配列と構造を生成するためのソフトウェア。 バイオインフォマティクス22,1534-1535. doi:10.1093/バイオインフォマティクス/btl113
PubMed要約/CrossRef全文/Google Scholar
Rho,M.,Tang,H.,Ye,Y.(2010). FragGeneScan:短くエラーが発生しやすい読み取りで遺伝子を予測する。 核酸Res.38、e191-e191。 土井:10.1093/nar/gkq747
PubMed要約/CrossRef全文/Google Scholar
Savoff,S.A.,Mitchell,A.J.,Schwaid,A.G.,Cabili,M.N.,Ma,J.,Levin,J.Z.,et al. (2013). ヒト細胞における短いオープンリーディングフレームコードされたペプチドのペプチド学的発見。 ナット ケム バイオル 9, 59–64. ドイ:10.1038/nchembio.1120
PubMed要約/CrossRef全文/Google Scholar
ることを可能にすることを目的としています。 (2018). リボソームプロファイリングデータの新規回帰分析によって明らかにされた保存された非AUG uORFs。 ゲノムRes.28,214-222. 土井:10.1101/gr.221507.117
Pubmed要約/CrossRef全文/Google Scholar
Vicens,Q.,Westhof,E.(2014). 環状Rnaの生合成。 セル159、13-14。 土井:10.1016/j。2014.09.005
PubMed要約/CrossRef全文/Google Scholar
Willems,P.,Ndah,E.,Jonckheere,V.,Stael,S.,Sticker,A.,Martens,l.,et al. (2017). N末端プロテオミクスは、シロイヌナズナの未踏の翻訳開始風景のプロファイリングを支援しました。 モル セル プロテオミクス16,1064-1080. ドイ:10.1074/mcp.M116.066662
PubMed要約/CrossRef全文/Google Scholar
Wu,J.,Mao,X.,Cai,T.,Luo,J.,Wei,L.(2006). KOBASサーバー:自動化された注釈および細道の同一証明のためのwebベースのプラットホーム。 核酸Res.34,W720–W724. doi:10.1093/nar/gkl167
PubMed要約/CrossRef全文/Google Scholar
Yang,L.,Fu,J.,Zhou,Y. (2018). 環状Rnaと免疫調節におけるそれらの新たな役割。 フロント イムノール 9, 2977. 土井:10.3389/fimmu.2018.02977
PubMed要約/CrossRef全文/Google Scholar
Yang,Y.,Fan,X.,Mao,M.,Song,X.,Wu,P.,Zhang,Y.,et al. (2017). N6-メチルアデノシンによって駆動される環状Rnaの広範な翻訳。 セルRes.27,626-641. doi:10.1038/cr。2017.31
PubMed要約|CrossRef全文|Google Scholar
Yu,G.,Wang,L.-G.,Han,Y.,He,Q.-Y.(2012). クラスタープロファイラー: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar