はじめに
四肢ガードル筋ジストロフィー(LGMDs)には、近位四肢ガードル筋の進行性消耗および弱さを特徴とする異種群が含まれる(1、2)。 LGMDsは、非常に軽度の形態から重度の早期発症、急速に進行する表現型に至るまで、家族内および家族内の変動性を表示する(2)。 LGMDは、常染色体優性(lgmd1)または劣性(lgmd2)に分類することができます。 最初のグループは通常、成人の発症年齢があり、あまり一般的ではありません(すべてのLGMDsの10%<)が、後者はより頻繁です(1:15000)(1)。 劣性型の中で、LGMD2A(またはカルパイン症)は、すべてのLgmd症例の約30%に影響を与える、世界的に最も一般的なLGMDである(3)。 疾患の臨床的特徴には、つま先歩行、歩行歩行、階段の走行および登山の困難、肩甲骨の翼が含まれる。 関節拘縮、アキレス腱短縮、脊柱側弯症が一般的に観察され、顔面および頸部の筋肉は影響を受けない(4)。 若年患者における無症候性高血症(5-80回CPK正常レベル)は、疾患の前臨床段階と考えられ、数年間持続することがあります(1、5)。 筋力低下の発症年齢は通常15歳で発生しますが、以前(<12歳)またはそれ以降(>30歳)の年齢で発生する可能性があります。 疾患の進行は、歩行の喪失、呼吸不全、および高度な段階での肺の生命能力の低下につながる可能性があります(6)。 心臓の関与(心臓律動障害、心臓伝導障害、左心室駆出機能不全)は時折しか報告されていない(6、7)。
カルパイン症の診断は、異なる代わりにスプライシングされた転写産物をコードするCAPN3(15q15.1)(4)の病原性変異の検出によって確認される。 しかし、全長転写物は、主に筋肉組織で発現される(8)。 エンコードされたタンパク質(CAPN3)は、非リソソームCa++依存性システインプロテアーゼファミリーのメンバーです。 筋肉では、CAPN3は機能および新陳代謝の要求に応じて筋肉適応そして成長のために必要である”サルコメアの改造”に加わります。 現在までに、CAPN3全体で490以上の病原性変異が記載されており、その大部分は一塩基変化である(4)。 CAPN3の変異は、ミトコンドリアの異常、成長障害、酸化ストレスの増加、および筋肉の解体と関連しており、筋肉が負荷を保持できなくなり、筋繊維の変性および筋肉の消耗を引き起こす原因となっている(8)。 カルパイン症の診断は、遺伝的異質性および臨床的および器械的パターンの非特異性のために挑戦することができる。 実際、筋力低下/萎縮、肥大/偽肥大、および腱拘縮の分布は、他のLgmdまたは神経筋障害と非常に頻繁に共有されています。 カルパイン症における筋生検パターンは、軽度の筋肉異常から重度のジストロフィー変化に至るまで、一般的に非特異的でもある。 また、免疫組織化学的/生化学的マーカーは、通常、信頼性がありません: カルパインシグナルは、非機能性タンパク質の存在下でも正常であり、その逆もまた、カルパイン症とは異なる他の筋ジストロフィーにおいても減少させることができる。
そのため、正確で信頼できる結果を得るために鑑別診断を行うことが推奨されます。 この目的のために、多遺伝子パネル、広範なゲノム(エクソーム/ゲノムシーケンシング)および単一遺伝子解析(直接配列決定)を含むカルパイ症の診断を確認するための分子遺伝学的検査アプローチが開発されている(9)。 次世代配列決定(NGS)の実施は、診断、予測または治療目的に適用される有益なデータを生成するのに有用であった(1 0−1 2)。 NGS遺伝子パネルは、遺伝的および表現型の異質性を特徴とする特定の疾患または関連障害の群に関連する遺伝子のセットの分析に基づいている。 NGSパネルは、親からの複数の遺伝的欠陥の遺伝に起因する混合または複雑な表現型を検出するためにも有用であり得る(13)。 一般に、複雑な表現型を提示する患者は、カスタム設計された診断遺伝子パネルの既知の突然変異に陰性であり、広範な鑑別診断を必要とする、全エキソームまたはゲノムシーケンシングの対象となる。 全エクソーム/ゲノムシーケンシングは、データ管理、解釈および分析コストの面でより高価なアプローチですが、疑われる遺伝性疾患(に分子診断を提供する可能性を増加させる13)。 LGMDsに関して、熱心なNGSのパネルは臨床テスト(9)の高い診断率、最適の適用範囲、感受性および特定性を保障するために強く推奨されています。 したがって、NGSパネルは、鑑別診断を容易にし、新しい原因突然変異を同定し、遺伝子型-表現型相関を明らかにするための最良のシステムの1つを表
この原稿では、LGMD2Aの影響を受けた患者の症例が報告されており、NGSパネルの適用により、カルパインパチー(CAPN3の変異)の診断を確認するだけでなく、拡張型心筋症に関連するLMNA遺伝子の追加の新規変異を同定することができた。 これらの結果を考えると、分析は、病理学的表現型に関連して分析データのより包括的な解釈を提供するために発端者の家族に拡張されました。
症例発表
発端者および親族の臨床的特徴
発端者における疾患の発症は、つま先歩行、ランニング、階段の登り、立ち上がらないふくらはぎの肥大の存在を報告した10歳の時に言及された。 症状は遅いが進行性の悪化を示し,その後近位上肢が関与した。 13歳の時、高レベルのCPK(〜8,000UI/L)が偶然に発見され、ミオグロビン尿症と関連することはありませんでした。 続いて、患者は様々な専門センターでいくつかの神経筋検査を受けた。 非特異的特徴を有する古典的なジストロフィー像(肥厚性/萎縮性線維、内部核、壊死性線維、結合組織の増加)を示す二つの筋肉生検を行った。 予想されるように、免疫化学およびimmunoblot研究は、壊死性線維においてのみα、β、γサルコグリカン、β-ジストログリカン、およびジストロフィンシグナルの異常/減少を示す決定的ではなかった(反復免疫化学はラミニンについても正常であった)。; ジストロフィンとカルパインに対する免疫ブロットは正常なシグナルを示した。 しかし、カルパイン-3immunoblotは、症例の20-30%が正常な量のタンパク質を示すので、LGMD2A診断には完全に感受性ではないことが知られている(1、16-18)。 臨床的提示(肥大症,ふくらはぎの肥大/偽肥大)によりジストロフィノパシーはまず除外された。 さらに、DMD(Xp21)、FKRP(19q13.32)、およびDYSF(2p13.2)遺伝子の分析は、病原性変異を明らかにしなかった。 患者は30歳で私たちの観察に来て、重要なwaddling歩行と翼の肩甲骨と、上肢と下肢の両方に軸方向とガードルの関与を提示しました。 下肢では、大腿四頭筋および大臀筋に衰弱が顕著であり、”明らかに肥大”(偽肥大)ふくらはぎの筋肉の証拠とともに有意に萎縮していた(図1)。 けいれん,筋痛,波紋は認められなかった。 完全なpneumological(肺活量測定およびpolysomnography)および心臓学的な(超音波検査、24時間ECGのHolterの監視、心臓MRI、目標とされた病歴および心血管の反射の分析の完全な心臓学的 我々はまた、酸マルターゼ欠乏症(リンパ球/白血球における正常な酵素活性アッセイ)を除外した。 筋磁気共鳴画像法(MRI)では、肩甲骨のガードル、傍脊椎筋、大腿部の後区画筋(サルトリウス筋とグラシリス筋の相対的な温存を伴う)、および脚(特に腓腹筋とヒラメウス)の顕著な関与が示された(図2)。 関与のこのパターンは、すでにカルパイン症(におけるMRI画像に関する文献で報告されている19、20)。 さらに、MRI研究では、ふくらはぎの筋肉が効果的に萎縮し、脂肪浸潤を特徴とし、筋肉の”拡大”を引き起こすことが示された。”臨床的提示、発症年齢、進行速度、筋力低下の分布および発端者におけるMRI所見は、文献(4、21、22)に広く記載されているLGMD2Aの診断と完全に一致していた。
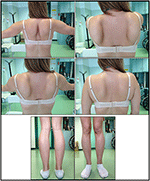
図1. 発端者の臨床的特徴:翼のある肩甲骨および大腿萎縮およびふくらはぎの偽肥大の組み合わせ。
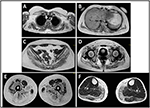
図2. 発端筋MRI: 上肢ガードルと傍脊椎筋の関与(A、B)、下肢ガードルと大腿の後区画(C–E、主にglutei)の関与、および脚の後区画(f、主に腓腹筋/ヒラメウス)の関与。
発端者の家族の評価では、神経筋疾患の病歴は明らかにされず、両親の間に血縁関係の証拠はなかった。 しかし、発端者の母親は、55歳で、特発性徐脈(夜までに40bpmまで)と診断された2つのsyncopalエピソードを提示した。 発端者の母親に対する広範な心臓学的調査は、いくつかのsyncopalエピソード/lipothymiaの発生後に症候性房室(AV)ブロックを診断した。 分析は、重度の徐脈のいくつかの期間と、基本的な第一度AVブロックの存在を示した、主に夜間としばしば症候性、第二度AVブロックのいくつかの相、1 心エコー検査およびエルゴメトリック検査では,卵円孔開存を除いて有意な変化は認められなかった。 母親は他の臨床症状、素因または危険因子を示さなかった。 彼女の臨床像を考えると、発端者の母親にPMKを移植した。 さらに、家族歴は、発端者の祖母と曾祖父もそれぞれ55歳と30歳でPMKを移植したことを明らかにした。 さらに、発端者の祖母の兄弟は、重度の拡張型心筋症のために51歳で死亡した。 心臓学的検査はまた、完全に影響を受けなかった発端者の父親と母親の叔父に対して行われた。
本研究は、Santa Lucia Foundationの倫理委員会によって承認され、ヘルシンキ宣言に従って実施された。 すべての参加者は、遺伝子解析のための署名されたインフォームドコンセントを提供し、この点で、彼らはまた、このケースレポートの公開のための同意を提
実験室の調査および診断テスト
ゲノムDNAは、MagPurix Blood DNA Extraction KitおよびMagPurix Automatic Extraction System(Resnova)を使用して、製造元の指示に従って末梢血(400μ l)から抽出しました。 試料は、Ion PGMシステムおよびIon Ampliseqカスタマイズパネル高特異性(Thermo Fisher Scientific)を使用して配列決定した。 パネルのサイズは129でした。13Kbで、パネル全体の99.72%を最小カバレッジで20倍にスクリーニングすることが期待されています。パネルには18個の遺伝子が含まれており、科学文献、GeneReviews(www.ncbi.nlm.nih.gov/books/NBK1116/)および一般集団における病原性変異体の頻度。 NGSパネルの詳細な説明を表1にまとめました。
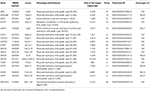
表1. LGMDの診断のために利用されるカスタマイズされたNGSのパネル。
ライブラリの構築は、Ion AmpliSeq™Library Kits2.0によって行われました。 約1 0ng/μ lの出発DNAを、多重PCR反応のために利用した。 連続的に、2つの精製工程(Ampure X P、Beckman Coulterを用いて)を行い、不要な汚染物質を除去し、最終PCRを行った。 テンプレート増幅は充実の手順を行ったIon PGMこんにちは-Q OT2キット-400、イオンOneTouch2システムおよびイオンOneTouch ES(サーモフィッシャー科学). サンプル加工によるイオンPGMこんにちは-Qシキット(400bp,サーモフィッシャー科学)、ライオン316チップv2(850-フローに必要なイオンPGMシーケンサー(サーモフィッシャー科学). 結果は、Ion Reporter4.6(Thermo Fisher Scientific)およびIntegrated Genome Viewer(IGV)を用いて分析した。 遺伝的変異体の解釈は、ヒト遺伝子変異データベース(HGMD)、ライデンオープンバリエーションデータベース(LOVD)、ClinvarおよびExacによって行われた。 検出された変異体の機能的効果は、変異テイスター、Varsome、SIFT、ポリフェン2、スマート、ヒトスプライシングファインダー(HSF)を含むバイオインフォマティック予測ツール ダイレクトシークエンシング(BigDyeターミネーター v3.遺伝子変異体を確認し、カバレッジ<1 0 1 1>2 0倍のゲノムコード領域を配列するために(LMNA,Chr1:1 5 6 1 0 6 0 5 2−1 5 6 1 0 6 0 7 6;DYSF,Chr2:7 1 7 5 3 3 5 2−7 1 7 5 3 5 0 2;Chr2:7 1 7 7 6 3 8 5−7 1 7 7 6 6 4 0;SGCB,Chr4:1 5 6 1 0 6 0 5 2−1 5 6 1 0 6 0 7 6;DYSF,Chr2:7 1 7 5 3 3 5 2−7 1 7 5 3 5 0 2;SGCB,Chr4:1 5 6 1 0 6 0 5 2−1 5 6 1 0 6 0 7 6;5 2 9 0 4 2 2 5−5 2 9 0 4 5 6 0;Sgca,chr1 7:4 8 2 4 3 2 4 2−4 8 2 4 3 5 7 0)。
結果
発端者のNGS分析では、3つのヘテロ接合変異体が明らかになった(補足図1)。 二つの変異体はCAPN3、すなわちNM_000070.2(CAPN3)に局在していた:c.550dela(P.Thr184Argfs)とc.1813G>c(P.Val605Leu)それぞれエクソン4と16。 最初は一塩基欠失であり、ヨーロッパ諸国で最も一般的な(症例の75%)病原性変異体である(1)。 予想されるように、バイオインフォマティック分析は、c.550dela(rs80338800)を機能喪失変異体(P.Thr184Argfs)として分類し、開いた読み取りフレームのフレームシフトを引き起こ 予測ツール(変異テイスター、HSF、Varsome、ポリフェン2、SIFT)は、変異体を有害であると説明した。 さらに、SMARTは、改変されたタンパク質生成物がカルパイン3ドメインと3つの「EF-hands」モチーフを欠いていることを明らかにした。 最初のドメインはカルパインのシグナル伝達経路に関与しているが、EF-handsはタンパク質のCa++依存性活性化に不可欠である(4)。
c.1813G>Cに関しては、スプライシングの潜在的な変化のために(突然変異Taster、HSF、Varsome、PolyPhen2、SIFTによって)有害であると予測されている新規ミスセンス変異体(p.Val605Leu)である。 この変異体は、文献またはオンラインデータベースの間で注釈されておらず、200の対照被験者では発見されていない。 残念なことに、SMART toolによる分析は、変異体が特徴的でないドメイン内に位置するため、有意な結果をもたらさなかった。 CAPN3の分離分析は、母親と母親の叔父の両方がc.550delaのヘテロ接合性であり、父親はc.1813G>C.
のキャリアであったことを示したさらに、発端者のNGS分析は、lmna、すなわちNM_170707(LMNA):c.550C>T(P.Gln184*)の新規変異体の存在を明らかにした。 過剰に言及された変異体はヌル変異体であると予測されており、文献またはオンラインデータベースにはまだ記載されておらず、200の対照被験者には見 予測ツール(突然変異テイスター、HSF、Varsome、ポリフェン2、SIFT)は、タンパク質産物に対するこの変異体の病原性効果を記述した。 スマートツールは、変更されたLMNAタンパク質は、その構造と機能を維持するために不可欠であるフィラメントドメインを欠いていることを報告した。 分離分析はcの存在を強調した。550C>t変異体は母親のみであり、彼女の心臓症状および彼女の家族歴との関連が示唆されている。
American College of Medical Genetics(ACMG)Standards and Guidelines(23)によって確立された基準によれば、c.1813G>C(CAPN3)は良性変異(PM1)のない重要なドメインに位置していることを考慮すると、病原性の可能性の高い変異体として記述することができる。ExAc、GnomAD、および1000ゲノムブラウザデータベース(PM2)には存在しない。; 遺伝の劣性モデルを考慮すると、それは病原性のある変異体(PM3)を有するtransで検出されている;計算証拠の複数の行は、遺伝子または遺伝子産物(PP3)に有害な影響をサポートしている。 ACMGによるc.550C>T(Lmna)の臨床分類に関しては、lmna(PVS1)における機能喪失を引き起こすヌルバリアント(P.Gln184*)であるため、病原性変異体として指定することができ、ExAc、GnomAD、1000ゲノムブラウザーデータベース(PM2)には存在しない。; 計算証拠の複数の行は、遺伝子または遺伝子産物(PP3)に対する有害な影響をサポートしています。
結論
この症例報告は、CAPN3(c.550delaおよびc.1813G>C)だけでなく、LMNA(c.550C>T)にも変異のキャリアであることが判明したLGMDの影響を受けた患者を提示した。 これらの結果は、capn3変異のために発端者の神経筋表現型を説明し、LMNAおよび彼女の陽性の家族歴における変異体の存在による心血管障害の潜在的 これらの結果を考慮して,家族を分離分析に供した。 特に、母はそれぞれCapn3_C.550delaおよびLmna_C.550C>Tのキャリアであることになった。 母親は、カルパイン症に関連するCapn3_C.550dela病原性変異の健康なキャリアと考えることができる。 しかし、LMNAにおける新規変異体の存在は、彼女の心血管病理(徐脈および失神エピソード)を説明する可能性がある。 母親における神経筋症候学の欠如および発端者の特異な臨床像は、Lmna_Cの可能性のある関連を除外した。550C>t神経筋表現型、特にEmery-Dreifuss筋ジストロフィー(EMD)に関する。 実際には、EMDは、特に比較的LGM2A(で免れるされている横広筋と上腕二頭筋に影響を与える24、25)。
母方の叔父はCapn3_C.550delaに対してのみヘテロ接合性であり、予想通り、神経筋または心血管の問題は示さなかった。 父親は小説Capn3_C.1813G>Cを運ぶことになり、それによって彼は影響を受けませんでした。 全体として、分離分析は、発端者の3つの突然変異の彼女の親戚からの遺伝を確認し、無視することができない心筋症の親しみやすさを強調した(図3)。
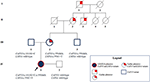
図3. 母系によって継承された心臓表現型と家族全体でCAPN3とLMNA変異体の伝達のための肯定的な親しみやすさを示す血統。
全体として、これらのデータはいくつかの重要な考慮事項を提起します。 まず、この患者のNGSパネルは、一つの既知の変異と病原性として予測される第二の新規変異体を検出し、カルパイン症の分子診断に到達するために重 第二に、NGSパネルは、発端者における新規Lmna_C.550C>T変異体の同定を可能にした。 この結果は、陽性の家族歴および分離データと一致し、母親の心血管疾患を説明し、さらに重要なことに、発端者のより具体的な心臓学的追跡を推奨した。 私たちは発端者がまだ”無症候性心臓病期”にある可能性があると仮定することができるように、心臓学的症状は、後の年齢(55歳)で母親に発生したこと「しかし、母親と比較して発端者におけるLMNA変異の可変表現型発現を排除することはできません。 これらのデータはすべて、結果の正確な解釈、適切な遺伝カウンセリング、そして最終的には臨床管理とフォローアップのために、臨床医と遺伝学者の間の統合されたアプローチの重要性を強調しています。 遺伝的および家族的カウンセリングに関しては,夫婦の生殖リスクを推定するために,発端者のパートナーについてもNGS分析を行った。 パートナーは、テストされた18のすべての遺伝子に陰性であり、テストが84%敏感であることを考慮すると、LGMD原因突然変異の健康なキャリアであるために1/650の残留リスクを有することを意味する。 発端者の遺伝的プロファイルとNGS検査の感受性を考慮すると、夫婦がカルパイン症に罹患した子供を持つ残留リスクは1/1300である。 一方、lmna病原性変異体は常染色体優性パターンに従って伝達され、発端者はヘテロ接合子を有する確率が50%であることを意味する。 しかし、lmna_C.550C>Tは新規な変異体であり、表現型に対するその機能的影響はまだ不明であるため、子孫の臨床像は確かに予測することはできません。
結論として、このケースレポートは、正確なLGMD2A診断を提供し、複数の遺伝子の異なる変異の遺伝に由来する複雑な表現型と併存疾患を記述するためのNGSパネルの臨床的有用性を強調している。 しかし、臨床診療におけるNGSの適用は、結果の明確な説明、患者の表現型に対する可能性のある意味、家族内の再発リスク、および予想外の所見を説明す
データの入手可能性
この研究のためにデータセットは生成または分析されませんでした。
倫理声明
この研究は、すべての被験者からの書面によるインフォームドコンセントを得て、サンタルチア財団の倫理委員会の勧告に従って行 すべての被験者は、ヘルシンキ宣言に従って書面によるインフォームドコンセントを与えた。 このプロトコルは、サンタルチア財団の倫理委員会によって承認されました。
著者の貢献
RC、CSt、VC、GC、RG、GPa、SC、CP、およびJMは、データの取得、分析、およびデータの解釈に貢献しました。 Cst、RC、GC、RG、GM、およびEGは原稿の起草に関与してきました。 SZ、GPr、CSa、およびSSは、臨床データの取得に関与してきた。 RC、CSt、VC、GC、RG、GPa、SC、CP、JM、SZ、GPr、GM、CSa、SS、およびEGは出版されるべき版の最終的な承認を与えました。
この作品は、2016年2月に厚生労働省によって支援されている。
利益相反声明
著者らは、この研究は利益相反の可能性と解釈される可能性のある商業的または財政的関係がない場合に行われたと宣言している。
補足資料
この記事の補足資料はオンラインで見つけることができます: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M、Angelini C.四肢ガードル型筋ジストロフィー2A型のタンパク質と遺伝子診断:収量と落とし穴。 筋肉神経。 (2015) 52:163–73. 土井:10.1002/mus.24682
PubMed要約/CrossRef全文/Google Scholar
2. Nigro V、Savarese M.四肢ガードル筋ジストロフィーの遺伝的基礎:2014年の更新。 アクタ-ミオル (2014) 33:1–12.
3. Richard I,Hogrel JY,Stockholm D,Payan CAM,Fougerousse F,Eymard B,et al. 臨床試験の結果の測定を描写するためのLGMD2Aの自然史。 アン-クリントランスロル… (2016) 3:248–65. ドイ:10.1002/acn3.287
PubMed要約/CrossRef全文/Google Scholar
4. Angelini C,Fanin M.Calpainopathy. 出演:アダムMP、アーディンガー HH、パゴンRA編集者。 ジェネレビューズ®。 ワシントン州シアトル:ワシントン大学(2017)1-30。
5. Kyriakides T,Angelini C,Schaefer J,Sacconi S,Siciliano G,Vilchez JJ,et al. Pauciへの診断アプローチに関するEFNSガイドライン-または無症候性高血症. ユール-ジュロル (2010) 17:767–73. ドイ:10.1111/j.1468-1331.2010.03012.x
CrossRef全文|Google Scholar
6. 森-吉村M,瀬川K,南N,大矢Y,小牧H,野中I,et al. 四肢ガードル型筋ジストロフィー患者の心肺機能障害2A.筋肉神経。 (2017) 55:465–9. 土井:10.1002/mus.25369
CrossRef全文|Google Scholar
7. Okere A、Reddy SS、Gupta S、Shinnar M.四肢ガードル型筋ジストロフィー2A型の患者における心筋症。Circ心臓が失敗する。 (2013)6:e12–13. 土井:10.1161/CIRCHEARTFAILURE.112.971424
CrossRef全文/Google Scholar
8. Kramerova I,Ermolova N,Eskin A,Hevener A,Quehenberger O,Armando AM,et al. 筋適応に必要な遺伝子の転写をアップレギュレートすることができないことは、四肢ガードル筋ジストロフィー2A(カルパイン症)の根底にある。 ハムモル-ジュネット (2016) 25:2194–207. doi:10.1093/hmg/ddw086
PubMed要約/CrossRef全文/Google Scholar
9. Thompson R,Straub V.Limb gardle muscular dystrophies-international collaborations for translational research. ナト-レヴロル (2016) 12:294–309. ドイ:10.1038/nrneurol.2016.35
PubMed要約/CrossRef全文|Google Scholar
10. Strafella C,Caputo V,Galota MR,Zampatti S,Marella G,Mauriello S,et al. 神経変性疾患における精密医療の応用。 前のページへ:前のページへ: (2018) 9:701. ドイ:10.3389/fneur.2018.00701
PubMed要約/CrossRef全文/Google Scholar
11. Cascella R,Strafella C,Caputo V,Errichiello V,Zampatti S,Milano F,et al. 加齢黄斑変性症における精密医療の応用に向けて。 ら(2 0 1 7)6 3:1 3 2−4 6. 土井:10.1016/j.preteyeres.2017.11.004
PubMed要約/CrossRef全文/Google Scholar
12. Cascella R,Strafella C,Longo G,Manzo L,Ragazzo M,De Felici C,et al. 遺伝カウンセリング、家族歴、および遺伝子検査を用いてAMDの個々のリスクを評価する。 アイ (2017) 32:446–50. ドイ:10.1038/目.2017.192
PubMed要約/CrossRef全文/Google Scholar
13. アダムス博士、Eng CM。 疑いのある遺伝性疾患を診断するための次世代シーケンシング。 N Engl J Med. (2019) 379:1353–62. doi:10.1056/Nejmc1814955
PubMed要約/CrossRef全文/Google Scholar
14. アンジェリーニC.神経筋疾患。 肢帯筋ジストロフィーにおける診断と発見。 ナト-レヴロル (2016) 12:6–8. ドイ:10.1038/nrneurol.2015.230
CrossRef全文/Google Scholar
15. Ghaoui R,Cooper ST,Lek M,Jones K,Corbett A,Reddel SW,et al. 四肢ガードル筋ジストロフィーの診断のための全エクソームシーケンシングの使用:結果と教訓を学んだ。 ジャマ-ノイロール (2015) 72:1424–32. ドイ:10.1001/ジャマヌロール.2015.2274
PubMed要約/CrossRef全文/Google Scholar
16. Milic A,Daniele N,Lochmüller H,Mora M,Comi GP,Moggio M,et al. LGMD2A生検の三分の一は、in vitroアッセイによって決定されるように、正常なカルパイン3タンパク質分解活性を有する。 ニューロムスク-ディゾルデ (2007) 17:148–56. 土井:10.1016/j.nmd.2006.11.001
PubMed抄録/CrossRef全文/Google Scholar
17. Lanzillo R,Aurino S,Fanin M,Aguennoz M,Vitale F,Fiorillo C,et al. 正常な非機能的なカルパイン3レベルの早期発症カルパイン症。 Dev Med子Neurol. (2006) 48:304–6. doi:10.1017/S001216220600065X
PubMed要約/CrossRef全文/Google Scholar
18. Talim B,Ognibene A,Mattioli E,Richard I,Anderson LV,Merlini L.遺伝的に確認された四肢ガードル型筋ジストロフィー2A型における正常なカルパイン発現。 (2001) 56:692–3. 土井:10.1212/wnl.56.5.692-a
CrossRef全文/Google Scholar
19. Díaz-Manera J,Llauger J,Gallardo E,Illa I.筋ジストロフィーにおける筋肉MRI. アクタ-ミオル (2015) 34:95–108.
20. Mercuri E,Bushby K,Ricci E,Birchall DR,Pane M,Kinali M,et al. カルパイン3欠乏症(LGMD2A)および早期拘縮を伴う四肢ガードル筋ジストロフィー患者における筋肉MRI所見。 ニューロムスク-ディゾルデ (2005) 15:164–71. 土井:10.1016/j.nmd.2004.10.008
PubMed抄録/CrossRef全文/Google Scholar
21. Magri F,Nigro V,Angelini C,Mongini T,Mora M,Moroni I,et al. イタリアの四肢ガードル筋ジストロフィーレジストリ: 相対的な頻度、臨床特徴および鑑別診断。 筋肉神経。 (2017) 55:55–68. 土井:10.1002/mus.25192
PubMed要約/CrossRef全文/Google Scholar
22. Chae J,Minami N,Jin Y,中川M,村山K,五十嵐F,et al. カルパイン3遺伝子変異:四肢ガードル筋ジストロフィーにおける遺伝的および臨床病理学的所見。 ニューロムスク-ディゾルデ (2001) 11:547–55. ドイ:10.1016/S0960-8966(01)00197-3
PubMed Abstract|CrossRef全文|Google Scholar
23. Richards S,Aziz N,Bale S,Bick D,Das S,Gastier-Foster J,et al. 配列変異体の解釈のための標準およびガイドライン:米国医学遺伝学およびゲノミクスの大学および分子病理学協会の共同コンセンサス勧告。 ジュネット-メド… (2015) 17:405–24. ドイ:10.1038/gim.2015.30
PubMed要約/CrossRef全文|Google Scholar
24. Bonne G,Mercuri E,Muchir A,Urtizberea A,Bécane HM,Recan D,et al. ラミンA/C遺伝子の変異による常染色体優性エメリー-Dreifuss筋ジストロフィーの臨床的および分子遺伝的スペクトル。 アン-ノイロール (2000) 48:170–80. 土井: 10.1002/1531-8249(200008)48:2<170::エイド-アナ6>3.0.CO;2-J
PubMed Abstract/CrossRef Full Text|Google Scholar
25. Hong JS,Ki CS,Kim JW,Suh YL,Kim JS,Baek KK,et al. Emery-Dreifuss筋ジストロフィーおよび四肢ガードル筋ジストロフィー1B型患者における心臓性不整脈、心筋症および筋ジストロフィー。J Korean Med Sci. (2005) 20:283–90. ドイ:10.3346/jkms.2005.20.2.283
クロスリファレンス全文|Google Scholar