Introduzione
Le Limb-Girdle Muscular Dystrophies (LGMDs) comprendono un gruppo eterogeneo di disturbi caratterizzati dal progressivo deperimento e debolezza dei muscoli prossimali degli arti (1, 2). Le LGMD mostrano variabilità inter e intrafamiliare, che vanno da forme molto lievi a fenotipi gravi, ad esordio precoce e rapidamente progressivi (2). Le LGMD possono essere classificate come autosomiche dominanti (LGMD1) o recessive (LGMD2). Il primo gruppo di solito ha un’età adulta di insorgenza e non sono molto comuni (<10% di tutte le LGMD), mentre i secondi sono più frequenti (1:15000) (1). Tra le forme recessive, la LGMD2A (o calpainopatia) è la LGMD più comune a livello mondiale, interessando circa il 30% di tutti i casi di LGMD (3). Le firme cliniche della malattia includono la camminata in punta di piedi, l’andatura ondeggiante, la difficoltà di correre e salire le scale, le ali scapolari. Si osservano comunemente contratture articolari, accorciamento del tendine di Achille, scoliosi, mentre i muscoli del viso e del collo non sono interessati (4). Un’iperckemia asintomatica (5-80 volte i livelli normali di CPK) nei pazienti giovani è considerata uno stadio preclinico della malattia e può persistere per diversi anni (1, 5). L’età di insorgenza della debolezza muscolare di solito si verifica a 15 anni, anche se può insorgere in età precedente (<12 anni) o successiva (>30 anni). La progressione della malattia può portare alla perdita di deambulazione, insufficienza respiratoria e ridotta capacità vitale polmonare negli stadi avanzati (6). Il coinvolgimento cardiaco (disturbi del ritmo cardiaco, disturbi della conduzione cardiaca, disfunzione di eiezione ventricolare sinistra) è riportato solo occasionalmente (6, 7).
La diagnosi di calpainopatia è confermata dalla rilevazione di mutazioni patogene in CAPN3 (15q15.1) (4), codificando diversi trascritti alternativamente impiombati. Tuttavia, la trascrizione integrale è espressa principalmente nel tessuto muscolare (8). La proteina codificata (CAPN3) è un membro della famiglia non lisosomiale Ca++-dipendente della cisteina proteasi. Nel muscolo, CAPN3 partecipa al “rimodellamento del sarcomero”, che è essenziale per l’adattamento e la crescita muscolare in risposta alle esigenze funzionali e metaboliche. Ad oggi, sono state descritte più di 490 mutazioni patogene in tutto CAPN3, la maggior parte delle quali sono cambiamenti a singolo nucleotide (4). Le mutazioni in CAPN3 sono state associate ad anomalie mitocondriali, fallimento della crescita, aumento dello stress ossidativo e disorganizzazione del sarcomero che complessivamente contribuiscono a rendere il muscolo incapace di sopportare carichi, causando quindi degenerazione della miofibra e deperimento muscolare (8). La diagnosi di calpainopatia può essere impegnativa a causa dell’eterogeneità genetica e della non specificità del pattern clinico e strumentale. Infatti, la distribuzione di debolezza muscolare / atrofia, ipertrofia / pseudo-ipertrofia e contratture tendinee sono molto spesso condivise con altri LGMD o disturbi neuromuscolari. Il modello di biopsia muscolare nella calpainopatia è generalmente non specifico, che va da lievi anomalie muscolari a gravi cambiamenti distrofici. Inoltre, i marcatori immunoistochimici / biochimici di solito non sono affidabili: il segnale calpain può essere normale anche in presenza di una proteina non funzionale, e viceversa può essere ridotto anche in altre distrofie muscolari diverse dalla calpainopatia.
Si raccomanda pertanto di eseguire una diagnosi differenziale al fine di fornire risultati accurati e affidabili. A questo scopo, sono stati sviluppati approcci di test genetici molecolari per confermare la diagnosi di calpainopatia, incluso il pannello multigene, l’analisi genomica estesa (sequenziamento dell’esoma/genoma) e l’analisi a singolo gene (sequenziamento diretto) (9). L’implementazione del sequenziamento di nuova generazione (NGS) è stata utile per generare dati informativi da applicare a scopi diagnostici, predittivi o terapeutici (10-12). I pannelli genetici NGS si basano sull’analisi di un insieme di geni associati a una specifica malattia o a un gruppo di disturbi correlati, che sono caratterizzati da eterogeneità genetica e fenotipica. I pannelli NGS possono anche essere utili per rilevare fenotipi misti o complessi, che derivano dall’ereditarietà di più di un difetto genetico da parte dei genitori (13). In generale, i pazienti che presentano fenotipi complessi, che sono negativi alle mutazioni note in pannelli genetici diagnostici progettati su misura e che richiedono un’ampia diagnosi differenziale, sono idonei per il sequenziamento dell’intero esoma o del genoma. Il sequenziamento dell’intero esoma / genoma è un approccio più costoso in termini di gestione dei dati, interpretazione e costi analitici, ma aumenta la probabilità di fornire una diagnosi molecolare a una sospetta malattia genetica (13). Per quanto riguarda le LGMD, i pannelli NGS dedicati sono altamente raccomandati, per garantire alti tassi diagnostici, copertura ottimale, sensibilità e specificità dei test clinici (9). I pannelli NGS rappresentano quindi uno dei migliori sistemi per facilitare la diagnosi differenziale, identificare nuove mutazioni causali e chiarire le correlazioni genotipo-fenotipo (14, 15).
In questo manoscritto viene riportato il caso di un paziente affetto da LGMD2A, per il quale l’applicazione del pannello NGS ha permesso non solo di confermare la diagnosi di calpainopatia (mutazioni in CAPN3) ma anche di identificare un’ulteriore, nuova mutazione nel gene LMNA associata a cardiomiopatia dilatativa. Alla luce di questi risultati, l’analisi è stata estesa ai membri della famiglia del probando per fornire un’interpretazione più completa dei dati analitici in relazione ai fenotipi patologici.
Presentazione del caso
Caratterizzazione clinica del Probando e dei parenti
L’insorgenza della malattia nel probando è stata riferita all’età di 10 anni, quando riporta la presenza di ipertrofia del polpaccio con camminata in punta di piedi, difficoltà nel correre, salire le scale e alzarsi in piedi. I sintomi hanno mostrato un peggioramento lento ma progressivo, con conseguente coinvolgimento dell’arto superiore prossimale. All’età di 13 anni, sono stati scoperti incidentalmente alti livelli di CPK (~8.000 UI/L), mai associati a mioglobinuria. Successivamente, il paziente è stato sottoposto a diverse indagini neuromuscolari in vari centri specializzati. Sono state eseguite due biopsie muscolari, mostrando il classico quadro distrofico (fibre ipertrofiche/atrofiche, nuclei interni, fibre necrotiche, aumento del tessuto connettivo) con caratteristiche non specifiche. Come previsto, gli studi di immunochimica e immunoblot erano inconcludenti che indicavano un segnale anormale/ridotto di α, β, γ sarcoglicano, β-distroglicano e distrofina solo nelle fibre necrotiche (l’immunochimica ripetuta risultava normale, anche per la laminina); immunoblots per distrofina e calpain hanno rivelato segnali normali. Tuttavia, l’immunoblot calpain-3 è noto per essere non completamente sensibile per la diagnosi LGMD2A, poiché il 20-30% dei casi mostra una quantità normale di proteine (1, 16-18). A causa della presentazione clinica (iperckmia, ipertrofia del vitello/pseudo-ipertrofia) le distrofinopatie sono state inizialmente escluse. Inoltre, l’analisi dei geni DMD (Xp21), FKRP (19q13.32) e DYSF (2p13.2) non ha rivelato mutazioni patogene. Il paziente è arrivato alla nostra osservazione all’età di 30 anni, presentando coinvolgimento assiale e cintola sia nell’arto superiore che in quello inferiore, con un’andatura ondulata significativa e scapole alate. Negli arti inferiori, la debolezza era prominente nei quadricipiti e nei glutei che erano significativamente ipo-atrofici, insieme all’evidenza di muscoli del polpaccio “apparentemente ipertrofici” (pseudo-ipertrofia) (Figura 1). Crampi, mialgie e increspature non sono stati osservati. Gli esami pneumologici completi (spirometria e polisonnografia) e cardiologici (ecografia, monitoraggio Holter ECG a 24 ore, risonanza magnetica cardiaca, ispezione fisica cardiologica completa con anamnesi mirata e analisi riflessa cardiovascolare) non hanno rivelato alcuna anomalia significativa. Abbiamo anche escluso il deficit di maltasi acida (normali saggi di attività enzimatica nei linfociti/leucociti). La risonanza magnetica muscolare (MRI) ha mostrato un coinvolgimento prominente del cingolo scapolare, dei muscoli paravertebrali, dei muscoli del compartimento posteriore della coscia (con relativo risparmio dei muscoli sartorius e gracilis) e della gamba (in particolare gastrocnemio mediale e soleo) (Figura 2). Questo modello di coinvolgimento è già stato riportato in letteratura riguardante il quadro RM in calpainopatia (19, 20). Inoltre, lo studio MRI ha dimostrato che i muscoli del polpaccio erano effettivamente atrofici e caratterizzati da infiltrazione di grasso, causando un “allargamento” muscolare.”La presentazione clinica, l’età di insorgenza, il tasso di progressione, la distribuzione della debolezza muscolare e i risultati della risonanza magnetica nel probando erano pienamente coerenti con una diagnosi di LGMD2A ampiamente descritta in letteratura (4, 21, 22).
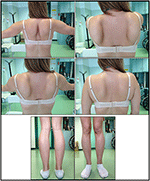
Figura 1. Caratteristiche cliniche del probando: scapole alate e combinazione di atrofia della coscia e pseudo-ipertrofia del polpaccio.
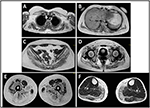
Figura 2. Muscolo probando MRI: coinvolgimento della cintura degli arti superiori e dei muscoli paravertebrali (A,B), coinvolgimento della cintura degli arti inferiori e del compartimento posteriore della coscia (C–E, principalmente i glutei) con relativo risparmio di sartorius e gracilis e coinvolgimento del compartimento posteriore della gamba (F, principalmente gastrocnemius medialis/soleus).
La valutazione dei membri della famiglia del probando non ha rivelato alcuna storia di malattia neuromuscolare e nessuna evidenza di consanguineità tra i genitori. Tuttavia, la madre del probando, all’età di 55 anni, ha presentato 2 episodi sincopali per i quali è stata diagnosticata una bradicardia idiopatica (fino a 40 bpm di notte). Ampie indagini cardiologiche sulla madre del probando hanno diagnosticato un blocco atrioventricolare sintomatico (AV) dopo il verificarsi di diversi episodi sincopali/lipotimia. L’analisi ha mostrato la presenza di un blocco AV di primo grado di base, con diversi periodi di grave bradicardia, per lo più notturna e spesso sintomatica, associata ad alcune fasi di blocco AV di secondo grado, sia di tipo 1 che 2, e alcune fasi di dissociazione AV completa notturna. L’ecocardiografia e il test ergometrico non hanno rivelato alterazioni significative, ad eccezione del forame ovale pervio. La madre non presentava alcun altro sintomo clinico, condizione predisponente o fattore di rischio. Dato il suo quadro clinico, la madre del probando è stata impiantata con PMK. Inoltre, la storia familiare ha rivelato che la nonna e il bisnonno del probando sono stati anche impiantati con PMK all’età di 55 e 30 anni, rispettivamente. Inoltre, il fratello della nonna del probando morì a 51 anni per una grave cardiomiopatia dilatativa. Sono stati eseguiti anche esami cardiologici sul padre e sullo zio materno del probando che sono risultati completamente inalterati.
Il presente studio è stato approvato dal comitato etico della Fondazione Santa Lucia ed è stato eseguito secondo la Dichiarazione di Helsinki. Tutti i partecipanti hanno fornito il consenso informato firmato per l’analisi genetica e, a questo proposito, hanno anche fornito il consenso per la pubblicazione di questo caso.
Indagini di laboratorio e test diagnostici
Il DNA genomico è stato estratto dal sangue periferico (400 µL) utilizzando MagPurix Blood DNA Extraction Kit e MagPurix Automatic Extraction System (Resnova) secondo le istruzioni del produttore. I campioni sono stati sequenziati utilizzando il sistema Ion PGM e il pannello personalizzato Ion Ampliseq ad alta specificità (Thermo Fisher Scientific). La dimensione del pannello era 129.13 Kb, che dovrebbe schermare ~99.72% del pannello totale con una copertura minima di 20X. Il pannello includeva 18 geni, che sono stati selezionati utilizzando la letteratura scientifica, GeneReviews (www.ncbi.nlm.nih.gov/books/NBK1116/) e frequenza delle varianti patogene nella popolazione generale. Una descrizione dettagliata del panel NGS è stata riassunta nella tabella 1.
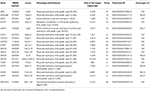
Tabella 1. Pannello su misura di NGS utilizzato per la diagnosi di LGMD.
La costruzione delle librerie è stata eseguita da Ion AmpliSeq™ Library Kits 2.0. Circa 10 ng/µl di DNA iniziale sono stati utilizzati per le reazioni di PCR multiplex. Successivamente, sono state eseguite due fasi di purificazione (utilizzando AMPure XP, Beckman Coulter) per rimuovere i contaminanti indesiderati ed è stata eseguita una PCR finale. Le fasi di amplificazione e arricchimento del modello sono state eseguite da Ion PGM Hi-Q OT2 kit-400, Ion OneTouch 2 System e Ion OneTouch ES (Thermo Fisher Scientific). I campioni sono stati elaborati da Ion PGM Hi-Q Sequencing Kit (400 bp, Thermo Fisher Scientific) ed eseguiti su Ion 316 Chip v2 (850 flussi richiesti) e Ion PGM Sequencer (Thermo Fisher Scientific). I risultati sono stati analizzati utilizzando Ion Reporter 4.6 (Thermo Fisher Scientific) e Integrated Genome Viewer (IGV). L’interpretazione delle varianti genetiche è stata condotta da Human Gene Mutation Database (HGMD), Leiden Open Variation Database (LOVD), ClinVar ed ExAC. L’effetto funzionale delle varianti rilevate è stato valutato da strumenti predittivi bioinformatici, tra cui Mutation Taster, Varsome, SIFT, PolyPhen 2, SMART, Human Splicing Finder (HSF). Sequenziamento diretto (Bigdye Terminator v3.1, BigDyeX Terminator e ABI3130, Applied Biosystems) è stata eseguita per confermare varianti genetiche e di sequenza genomica di codifica regioni con una copertura <20X (LMNA, Chr1:156106052-156106076; DYSF, Chr2:71753352-71753502, Chr2:71776385-71776640; SGCB, Chr4:52904225-52904560; SGCA, Chr17:48243242-48243570).
Risultati
L’analisi NGS del probando ha rivelato 3 varianti eterozigote (Figura supplementare 1). Due varianti sono state localizzate in CAPN3, vale a dire NM_000070.2 (CAPN3): c.550delA (p.Thr184Argfs) e c.1813G>C (p.Val605Leu) negli esoni 4 e 16, rispettivamente. La prima è una delezione a singolo nucleotide ed è la variante patogena più comune (75% dei casi) nei Paesi europei (1). Come previsto, l’analisi bioinformatica ha classificato il c.550delA (rs80338800) come variante di perdita di funzione (p.Thr184Argfs), causando un frameshift del frame di lettura aperto. Gli strumenti predittivi (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) hanno descritto la variante come deleteria. Inoltre, SMART ha rivelato che il prodotto proteico alterato manca del dominio calpain 3 e dei tre motivi “EF-hands”. Il primo dominio è coinvolto nelle vie di segnalazione di Calpain mentre le mani EF sono essenziali per l’attivazione Ca++-dipendente della proteina (4).
Per quanto riguarda c.1813G > C, è una nuova variante missense (p.Val605Leu) che è stata predetta come deleteria (da Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) a causa di una potenziale alterazione dello splicing. Questa variante non è annotata in letteratura o tra i database online e non è stata trovata in 200 soggetti di controllo. Sfortunatamente, l’analisi di SMART tool non ha prodotto risultati significativi, poiché la variante si trova all’interno di un dominio non caratteristico. L’analisi di segregazione di CAPN3 ha mostrato che la madre e lo zio materno erano entrambi eterozigoti per c.550delA, mentre il padre era portatore di c.1813G>C.
Inoltre, l’analisi NGS sul probando ha rivelato la presenza di una nuova variante in LMNA, vale a dire NM_170707 (LMNA): c.550C>T (p.Gln184*). La variante sopra menzionata è stata prevista come una variante nulla, non è stata ancora descritta in letteratura o tra i database online e non è stata trovata in 200 soggetti di controllo. Gli strumenti predittivi (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) hanno descritto un effetto patogeno di questa variante sul prodotto proteico. SMART tool ha riferito che la proteina LMNA alterata manca del dominio del filamento, che è essenziale per mantenere la sua struttura e funzione. L’analisi di segregazione ha evidenziato la presenza del c.550C > T variante solo nella madre, suggerendo una possibile associazione con la sua sintomatologia cardiaca e la sua storia familiare di malattia.
Secondo i criteri stabiliti dagli standard e dalle linee guida dell’American College of Medical Genetics (ACMG) (23), c. 1813G > C (CAPN3) può essere descritto come una probabile variante patogena considerando che si trova in un dominio critico senza variazione benigna (PM1); è assente nei database EXAC, GnomAD e 1000 Genome Browser (PM2).; considerando il modello recessivo di ereditarietà, è stato rilevato in trans con una variante patogena (PM3); più linee di evidenza computazionale supportano un effetto deleterio sul gene o sul prodotto genico (PP3). Per quanto riguarda la classificazione clinica di c. 550C > T (LMNA) di ACMG, può essere designato come variante patogena, poiché è una variante nulla che causa perdita di funzione (p.Gln184*) in LMNA (PVS1); è assente nei database EXAC, GnomAD e 1000 Genome Browser (PM2); si trova in un dominio critico senza variazione benigna (PM1); linee multiple di prove computazionali supportano un effetto deleterio sul gene o prodotto genico (PP3).
Conclusioni
Questo rapporto di caso ha presentato un paziente affetto da LGMD, che è risultato essere portatore di mutazioni non solo in CAPN3 (c.550delA e c.1813G>C) ma anche in LMNA (c.550C > T). Questi risultati spiegano il fenotipo neuromuscolare del probando a causa delle mutazioni di CAPN3 ed evidenziano un potenziale rischio di disturbi cardiovascolari a causa della presenza della variante in LMNA e della sua storia familiare positiva. Alla luce di questi risultati, i membri della famiglia sono stati sottoposti all’analisi di segregazione. In particolare, la madre risultava essere portatrice di CAPN3_c.550delA e LMNA_c.550C>T, rispettivamente. La madre può essere considerata come portatrice sana della mutazione patogena CAPN3_c.550delA associata alla calpainopatia. Tuttavia, la presenza di una nuova variante in LMNA può spiegare la sua patologia cardiovascolare (bradicardia e episodi sincopali). L’assenza di sintomatologia neuromuscolare nella madre e il particolare quadro clinico del probando escludevano una possibile associazione di LMNA_c.550C> T con fenotipo neuromuscolare, in particolare per quanto riguarda la distrofia muscolare di Emery-Dreifuss (EMD). Infatti, l’EMD colpisce specificamente i muscoli del vasto lateralis e del bicipite brachiale che sono relativamente risparmiati in LGM2A (24, 25).
Lo zio materno era eterozigote solo per il CAPN3_c.550delA e, come previsto, non presentava problemi neuromuscolari o cardiovascolari. Il padre ha portato a portare il romanzo CAPN3_c. 1813G > C e, quindi, non è stato influenzato. Nel complesso, l’analisi della segregazione ha confermato l’eredità delle tre mutazioni del probando dai suoi parenti e ha evidenziato una familiarità per la cardiomiopatia che non può essere trascurata (Figura 3).
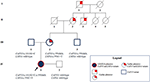
Figura 3. Pedigree che mostra la familiarità positiva per il fenotipo cardiaco ereditato dal lignaggio materno e la trasmissione delle varianti CAPN3 e LMNA in tutti i membri della famiglia.
Complessivamente, questi dati sollevano alcune considerazioni importanti. In primo luogo, il pannello NGS in questo paziente è stato fondamentale per raggiungere la diagnosi molecolare della calpainopatia, rilevando una mutazione nota e una seconda nuova variante predetta come patogena. In secondo luogo, il pannello NGS ha permesso l’identificazione della nuova variante LMNA_c.550C>T nel probando. Questo risultato era coerente con la storia familiare positiva e con i dati di segregazione, spiegando le malattie cardiovascolari nella madre e, cosa più importante, raccomandando un follow-up cardiologico più specifico nel probando. È importante notare che le manifestazioni cardiologiche si sono verificate nella madre più tardi di età (55 anni), in modo che possiamo supporre che il probando possa ancora essere in una “fase cardiologica asintomatica.”Tuttavia, un’espressione fenotipica variabile della mutazione LMNA nel probando rispetto alla madre non può essere esclusa. Tutti questi dati sottolineano l’importanza di un approccio integrato tra clinici e genetisti, per una corretta interpretazione dei risultati, una corretta consulenza genetica e, eventualmente, gestione clinica e follow-up. Per quanto riguarda la consulenza genetica e familiare, l’analisi NGS è stata eseguita anche sul partner del probando, al fine di stimare il rischio riproduttivo per la coppia. Il partner è risultato negativo a tutti i 18 geni testati, il che significa che ha un rischio residuo di 1/650 di essere un portatore sano per le mutazioni causali LGMD, considerando che il test è sensibile all ‘ 84%. Prendendo in considerazione il profilo genetico del probando e la sensibilità del test NGS, il rischio residuo per la coppia di avere un figlio affetto da calpainopatia è 1/1300. D’altra parte, le varianti patogene LMNA sono trasmesse secondo uno schema autosomico dominante, il che significa che il probando ha una probabilità del 50% di avere figli eterozigoti. Tuttavia, il quadro clinico della prole non può essere certamente previsto in quanto LMNA_c.550C > T è una nuova variante e il suo impatto funzionale sul fenotipo è ancora sconosciuto.
In conclusione, questo case report evidenzia l’utilità clinica dei pannelli NGS per fornire una diagnosi accurata di LGMD2A e descrivere fenotipi complessi e comorbidità derivanti dall’ereditarietà di diverse mutazioni in più geni. Tuttavia, l’applicazione di NGS nella pratica clinica deve sempre essere combinata con una consulenza pre e post-genetica al fine di fornire una chiara spiegazione dei risultati, le possibili implicazioni sul fenotipo dei pazienti, il rischio di recidiva all’interno della famiglia, nonché per spiegare possibili risultati inaspettati.
Disponibilità dei dati
Non sono stati generati o analizzati set di dati per questo studio.
Dichiarazione etica
Questo studio è stato condotto in conformità con le raccomandazioni del comitato etico della Fondazione Santa Lucia con il consenso informato scritto di tutti i soggetti. Tutti i soggetti hanno dato il consenso informato scritto in conformità con la Dichiarazione di Helsinki. Il protocollo è stato approvato dal comitato etico della Fondazione Santa Lucia.
Contributi dell’autore
RC, CSt, VC, GC, RG, GPa, SC, CP e JM hanno contribuito all’acquisizione dei dati, all’analisi e all’interpretazione dei dati. CSt, RC, GC, RG, GM, e EG sono stati coinvolti nella stesura del manoscritto. SZ, GPr, CSa e SS sono stati coinvolti nell’acquisizione di dati clinici. RC, CSt, VC, GC, RG, GPa, SC, CP, JM, SZ, GPr, GM, CSa, SS e EG hanno dato l’approvazione finale della versione da pubblicare.
Finanziamento
Questo lavoro è sostenuto da 5X 2016 Ministero della Salute Nazionale 2.
Dichiarazione sul conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.
Materiale supplementare
Il materiale supplementare per questo articolo può essere trovato online all’indirizzo: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. Proteina e diagnosi genetica della distrofia muscolare di tipo 2A: la resa e le insidie. Nervo muscolare. (2015) 52:163–73. doi: 10.1002 / mus.24682
PubMed Abstract / CrossRef Testo completo / Google Scholar
2. Nigro V, Savarese M. Basi genetiche delle distrofie muscolari arto-cingolo: l’aggiornamento 2014. Acta Myol. (2014) 33:1–12.
PubMed Abstract / Google Scholar
3. Richard I, Hogrel JY, Stockholm D, Payan CAM, Fougerousse F, Eymard B, et al. Storia naturale di LGMD2A per delineare le misure di outcome negli studi clinici. Ann Clin Transl Neurol. (2016) 3:248–65. doi: 10.1002 / acn3.287
PubMed Abstract / CrossRef Testo completo / Google Scholar
4. Angelini C, Fanin M. Calpainopatia. In: Adam MP, Ardinger HH, Pagon RA editori. GeneReviews®. Il sito è stato aperto nel 2017.
PubMed Abstract / Google Scholar
5. Kyriakides T, Angelini C, Schaefer J, Sacconi S, Siciliano G, Vilchez JJ, et al. Linee guida EFNS sull’approccio diagnostico all’iperckemia pauci – o asintomatica. Eur J Neurol. (2010) 17:767–73. doi: 10.1111 / j. 1468-1331. 2010.03012.x
Testo completo CrossRef / Google Scholar
6. Il suo nome deriva da quello di una donna di nome Yoshimura. Disfunzione cardiopolmonare in pazienti con distrofia muscolare della cintura degli arti 2A. Nervo muscolare. (2017) 55:465–9. doi: 10.1002 / mus.25369
CrossRef Testo completo / Google Scholar
7. Okere A, Reddy SS, Gupta S, Shinnar M. A cardiomiopatia in un paziente con arto cintura distrofia muscolare di tipo 2A. Cuore Circ fallire. (2013)6: e12-13. doi: 10.1161 / CIRCHEARTFAILURE.112.971424
CrossRef Testo completo / Google Scholar
8. Kramerova I, Ermolova N, Eskin A, Hevener A, Quehenberger O, Armando AM, et al. La mancata up-regolare la trascrizione dei geni necessari per l’adattamento muscolare è alla base della distrofia muscolare della cintura degli arti 2A (calpainopatia). Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093/hmg / ddw086
PubMed Abstract / CrossRef Testo completo / Google Scholar
9. Thompson R, Straub V. Limb girdle muscular dystrophies-collaborazioni internazionali per la ricerca traslazionale. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038 / nrneurol.2016.35
PubMed Abstract / CrossRef Testo completo / Google Scholar
10. Strafella C, Caputo V, Galota MR, Zampatti S, Marella G, Mauriello S, et al. Applicazione della medicina di precisione nelle malattie neurodegenerative. Neurolo anteriore. (2018) 9:701. doi: 10.3389 / fneur.2018.00701
PubMed Abstract | CrossRef Testo completo / Google Scholar
11. Cascella R, Strafella C, Caputo V, Errichiello V, Zampatti S, Milano F, et al. Verso l’applicazione della medicina di precisione nella degenerazione maculare legata all’età. Prog Retin Eye Res. (2017) 63:132-46. doi: 10.1016 / j. preteyeres.2017.11.004
PubMed Abstract | CrossRef Testo completo / Google Scholar
12. Cascella R, Strafella C, Longo G, Manzo L, Ragazzo M, De Felici C, et al. Valutazione del rischio individuale per AMD con consulenza genetica, storia familiare e test genetici. Occhio. (2017) 32:446–50. doi: 10.1038 / occhio.2017.192
PubMed Abstract / CrossRef Testo completo / Google Scholar
13. Adams DR, Ing CM. Sequenziamento di nuova generazione per diagnosticare malattie genetiche sospette. N Ingl J Med. (2019) 379:1353–62. doi: 10.1056 / NEJMc1814955
PubMed Abstract / CrossRef Testo completo / Google Scholar
14. Angelini C. Malattia neuromuscolare. Diagnosi e scoperta nella distrofia muscolare degli arti. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038 / nrneurol.2015.230
CrossRef Testo completo / Google Scholar
15. Il sito utilizza cookie tecnici e di terze parti. Uso del sequenziamento dell’intero esoma per la diagnosi della distrofia muscolare arto-cintura: risultati e lezioni apprese. JAMA Neurol. (2015) 72:1424–32. doi: 10.1001 / jamaneurol.2015.2274
PubMed Abstract / CrossRef Testo completo / Google Scholar
16. Milic A, Daniele N, Lochmüller H, Mora M, Comi GP, Moggio M, et al. Un terzo delle biopsie LGMD2A ha una normale attività proteolitica calpain 3 determinata da un test in vitro. Disturbo neuromusc. (2007) 17:148–56. doi: 10.1016 / j. nmd.2006.11.001
PubMed Abstract | CrossRef Full Text / Google Scholar
17. Lanzillo R, Aurino S, Fanin M, Aguennoz M, Vitale F, Fiorillo C, et al. Calpainopatia ad esordio precoce con normale livello di calpain 3 non funzionale. Dev Med Neurol bambino. (2006) 48:304–6. doi: 10.1017 / S001216220600065X
PubMed Abstract / CrossRef Full Text / Google Scholar
18. Talim B, Ognibene A, Mattioli E, Richard I, Anderson LV, Merlini L. Normale espressione calpain nella distrofia muscolare geneticamente confermata tipo 2A. Neurologia. (2001) 56:692–3. doi: 10.1212 / wnl.56.5.692-a
CrossRef Testo completo / Google Scholar
19. Díaz-Manera J, Llauger J, Gallardo E, MRI I. Risonanza magnetica muscolare nelle distrofie muscolari. Acta Myol. (2015) 34:95–108.
PubMed Abstract / Google Scholar
20. Mercuri E, Bushby K, Ricci E, Birchall DR, Pane M, Kinali M, et al. Risultati della risonanza magnetica muscolare in pazienti con distrofia muscolare della cintura degli arti con deficit di calpain 3 (LGMD2A) e contratture precoci. Disturbo neuromusc. (2005) 15:164–71. doi: 10.1016 / j. nmd.2004.10.008
PubMed Abstract | CrossRef Testo completo / Google Scholar
21. Il suo nome deriva dal greco antico, che significa “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”. Il registro italiano della distrofia muscolare della cintura degli arti: frequenza relativa, caratteristiche cliniche e diagnosi differenziale. Nervo muscolare. (2017) 55:55–68. doi: 10.1002 / mus.25192
PubMed Abstract / CrossRef Testo completo / Google Scholar
22. Il suo nome deriva dal greco antico, che significa “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra” e “terra”. Calpain 3 mutazioni genetiche: scoperte genetiche e clinico-patologiche nella distrofia muscolare arto-cingolo. Disturbo neuromusc. (2001) 11:547–55. doi: 10.1016 / S0960-8966(01)00197-3
PubMed Abstract / CrossRef Testo completo / Google Scholar
23. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standard e linee guida per l’interpretazione delle varianti di sequenza: una raccomandazione di consenso congiunto dell’American College of Medical Genetics and Genomics e dell’Associazione per la patologia molecolare. Genet Med. (2015) 17:405–24. doi: 10.1038 / gim.2015.30
PubMed Abstract / CrossRef Testo completo / Google Scholar
24. Bonne G, Mercuri E, Muchir A, Urtizberea A, Bécane HM, Recan D, et al. Spettro genetico clinico e molecolare della distrofia muscolare autosomica dominante Emery-Dreifuss dovuta a mutazioni del gene lamin A/C. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170::AIUTI-ANA6>3.0.CO; 2-J
PubMed Abstract / CrossRef Full Text / Google Scholar
25. Il suo nome deriva dal latino “Hong JS”, “Ki CS”, “Kim JW”, “Suh YL”, “Kim JS”, “Baek KK”, et al. Disritmie cardiache, cardiomiopatia e distrofia muscolare in pazienti con distrofia muscolare Emery-Dreifuss e distrofia muscolare degli arti-cintura tipo 1B. J Coreano Med Sci. (2005) 20:283–90. doi: 10.3346 / jkms.2005.20.2.283
CrossRef Full Text / Google Scholar