- Introduzione
- Metodi
- Filtraggio dei dati di profilazione ribosomiale
- Assemblaggio di genomi virtuali
- Determinazione della regione Read-Mapped di Ribo-seq su una giunzione (RMRJ) di circRNAs
- Formazione del modello e classificazione di RMRJs
- La previsione dei peptidi tradotti da RMRJs
- Risultati e discussione
- circRNAs tradotto negli esseri umani e A. thaliana
- Arricchimento funzionale di circRNAs umani e A. thaliana con potenziale di codifica
- Test di Precisione per CircCode
- Influenza della profondità di sequenziamento dei dati Ribo-seq
- Confronto del CircCode con altri strumenti
- Conclusioni
- Disponibilità e Requisiti
- Dichiarazione di disponibilità dei dati
- Contributi dell’autore
- Finanziamento
- Conflitto di interessi
- Materiale supplementare
Introduzione
Gli RNA circolari (CIRCRNA) sono un tipo speciale di molecola di RNA non codificante che è diventato un argomento di ricerca caldo nel campo dell’RNA e sta ricevendo molta attenzione (Chen e Yang, 2015). Rispetto agli RNA lineari tradizionali (contenenti estremità 5′ e 3′), le molecole di circRNA di solito hanno una struttura circolare chiusa; rendendole più stabili e meno inclini alla degradazione (Vicens e Westhof, 2014). Sebbene l’esistenza di CIRCRNA sia nota da tempo, queste molecole sono state considerate un sottoprodotto dello splicing dell’RNA. Tuttavia, con lo sviluppo di tecnologie di sequenziamento e bioinformatica ad alto rendimento, i CIRCRNA sono diventati ampiamente riconosciuti negli animali e nelle piante (Chen e Yang, 2015). Studi recenti hanno anche dimostrato che un gran numero di CIRCRNA può essere tradotto in piccoli peptidi nelle cellule (Pamudurti et al., 2017) e hanno ruoli chiave nonostante il loro livello di espressione a volte basso (Hsu e Benfey, 2018; Yang et al., 2018). Sebbene si stia identificando un numero crescente di CIRCRNA, le loro funzioni nelle piante e negli animali rimangono generalmente da studiare. Oltre alle loro funzioni come richiami di miRNA, i CIRRNA hanno un importante potenziale traslazionale, ma non sono disponibili strumenti per prevedere specificamente le capacità traslazionali di queste molecole (Jakobi e Dieterich, 2019).
Esistono diversi strumenti per la previsione e l’identificazione dei CIRRI, come CIRI (Gao et al., 2015), CIRCexplorer (Dong et al., 2019), CircPro (Meng et al., 2017), e circtools (Jakobi et al., 2018). Tra questi, CircPro può rivelare i CIRCRNA tradotti calcolando un punteggio potenziale di traduzione per i CIRCRNA basato su CPC (Kong et al., 2007), che è uno strumento per identificare il frame di lettura aperto (ORF) in una data sequenza. Tuttavia, poiché alcuni CIRCRNA non usano il codone iniziale durante la traduzione (Ingolia et al., 2011; Slavoff et al., 2013; Kearse e Wilusz, 2017; Spealman et al., 2018), impiegando CPC può filtrare alcuni circRNAs veramente tradotti. In questo studio, abbiamo usato BASiNET (et et al., 2018), che è un classificatore di RNA basato sui metodi di apprendimento automatico (foresta casuale e modello J48). Inizialmente trasforma gli RNA di codifica dati (dati positivi) e gli RNA non codificanti (dati negativi) e li rappresenta come reti complesse; estrae quindi le misure topologiche di queste reti e costruisce un vettore di funzionalità per addestrare il modello che viene utilizzato per classificare la capacità di codifica dei CIRCRNA. Con questo metodo, viene evitato un filtraggio errato dei circRNAs tradotti che non sono iniziati da AUG. Inoltre, la tecnologia Ribo-seq, che si basa sul sequenziamento ad alto throughput per monitorare RPFs (frammenti protetti ribosomiali) di trascrizioni (Guttman et al., 2013; Brar e Weissman, 2015), può essere utilizzato per determinare le posizioni dei CIRCRNA che vengono tradotti (Michel e Baranov, 2013). Per identificare la capacità di codifica di CIRCRNA, abbiamo sviluppato lo strumento CircCode, che coinvolge un framework basato su Python 3, e applicato CircCode per indagare il potenziale di traduzione di CIRCRNA da esseri umani e Arabidopsis thaliana. Il nostro lavoro fornisce una ricca risorsa per ulteriori studi sulle funzioni dei CIRCRNA con capacità di codifica.
Metodi
CircCode è stato scritto nel linguaggio di programmazione Python 3; utilizza Trimmomatic (Bolger et al. Nel 2014, bowtie (Langmead and Salzberg, 2012) e STAR (Dobin et al., 2013) per filtrare raw Ribo-seq legge e mappare queste letture filtrate al genoma. CircCode identifica quindi le regioni mappate in lettura Ribo-seq in circRNAs che contengono giunzioni. Successivamente, le sequenze mappate candidate nei CIRCRNA vengono ordinate in base ai classificatori (modello J48) in RNA di codifica e RNA non codificanti da BASiNET. Infine, i peptidi brevi prodotti dalla traduzione sono identificati come potenziali regioni codificanti dei CIRCRNA. L’intero processo di CircCode consiste di cinque passaggi (Figura 1).
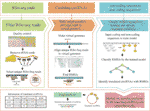
Figura 1 Il flusso di lavoro di CircCode. Il livello superiore rappresenta il file di input richiesto per ogni passaggio di CircCode. Lo strato intermedio è diviso in tre parti e ogni parte rappresenta una diversa fase di funzionamento. Da sinistra a destra, la prima parte rappresenta il filtraggio dei dati Ribo-seq; il controllo di qualità viene eseguito da Trimmomatic e le letture rRNA vengono rimosse da bowtie. La seconda parte rappresenta i passaggi utilizzati per produrre il genoma virtuale e allineare le letture filtrate al genoma virtuale con STAR. L’ultima parte rappresenta l’identificazione dei circRNAs tradotti dall’apprendimento automatico. Lo strato inferiore rappresenta l’ultimo passaggio utilizzato per prevedere i peptidi tradotti dai CIRCRNA e i risultati finali dell’output, comprese le informazioni sui CIRCRNA tradotti e sui loro prodotti di traduzione.
Filtraggio dei dati di profilazione ribosomiale
In primo luogo, frammenti e adattatori di bassa qualità nelle letture Ribo-Seq vengono rimossi da Trimmomatic con i parametri predefiniti per ottenere letture Ribo-seq pulite. In secondo luogo, queste letture pulite di Ribo-seq sono mappate su una libreria rRNA per rimuovere le letture derivate da rRNA usando bowtie. Poiché le lunghezze di lettura di Ribo-seq sono relativamente brevi (generalmente inferiori a 50 bp), è possibile che una lettura corrisponda a più regioni. In questo caso, è difficile determinare a quale regione corrisponde una particolare lettura. Per evitare ciò, le letture pulite di Ribo-seq sono mappate sul genoma di una specie di interesse e le letture che non sono perfettamente allineate al genoma sono considerate come le letture uniche finali di Ribo-seq.
Assemblaggio di genomi virtuali
I CIRRNA di solito appaiono come molecole a forma di anello negli eucarioti e possono essere identificati in base alle loro giunzioni di back-splicing. Tuttavia, le sequenze di CIRCRNA nel file fasta sono spesso in forma lineare. In teoria, il risultato indica che la giunzione è tra il nucleotide terminale 5′ e il nucleotide terminale 3′, sebbene la giunzione e la sequenza vicino alla giunzione non possano essere visualizzate direttamente, allineando così le letture Ribo-seq alle sequenze circRNA, comprese le giunzioni, in modo semplice.
CircCode collega la sequenza di ciascun circRNA in tandem in modo tale che la giunzione per ciascuno si trovi nel mezzo della sequenza di nuova costruzione. Abbiamo anche separato ogni unità di serie di 100 nucleotidi N per evitare confusione nella fase di allineamento della sequenza (la lunghezza di ciascun RPF è inferiore a 50 bp). Infine, abbiamo ottenuto un genoma virtuale costituito solo da CIRCRNA candidati in tandem separati da 100 Ns. Poiché CircCode si concentra solo sull’allineamento tra le letture Ribo-seq e le sequenze circRNA, possiamo studiare il potenziale di codifica dei CIRCRNA mappando le letture Ribo-seq a questo genoma virtuale, che può risparmiare una grande quantità di tempo computazionale (il genoma virtuale è molto più piccolo dell’intero genoma) e aumentare la precisione (evitando interferenze tra i confronti delle sequenze upstream e downstream dei CIRCRNA).
Determinazione della regione Read-Mapped di Ribo-seq su una giunzione (RMRJ) di circRNAs
Le letture uniche finali di Ribo-seq sono mappate su un genoma virtuale creato in precedenza utilizzando STAR. Poiché ogni unità CIRCRNA tandem è stato separato da 100 basi N prima di produrre il genoma virtuale, la più grande lunghezza introne è stato impostato per non superare 10 basi con il parametro “–alignIntronMax 10.”Questo parametro elimina qualsiasi interazione tra diversi CIRCRNA nell’allineamento della sequenza. Nella seconda fase della produzione del genoma virtuale, CircCode memorizza le informazioni di giunzione posizionale per ciascun circRNA nel genoma virtuale. Se la regione read-mapped Ribo-seq nel genoma virtuale include la giunzione del circRNA e il numero di letture mappate Ribo-seq sulla giunzione (NMJ) è maggiore di 3, la regione reads-mapped Ribo-seq sulla giunzione dei CIRCRNA può essere considerata come un RMRJ, che rivela un segmento approssimativamente tradotto di CIRCRNA vicino al sito di giunzione.
Formazione del modello e classificazione di RMRJs
Sebbene RMRJs possa costituire una potente prova di traduzione, ci sono ancora alcune carenze in questo metodo. Poiché la lunghezza delle letture della mappa ribosomiale è breve, una lettura può essere paragonata alla posizione sbagliata. Pertanto, non è convincente considerare semplicemente la regione coperta dalla legge Ribo-seq come la regione tradotta. A tal fine, il metodo di apprendimento automatico viene utilizzato per identificare la capacità di codifica del RMRJ. In primo luogo, CircCode estrae gli RNA codificanti (dati positivi) e gli RNA non codificanti (dati negativi) da una specie di interesse e li utilizza per l’addestramento del modello mediante la differenza di vettori di funzionalità tra RNA codificanti e non codificanti. CircCode utilizza quindi il modello addestrato per classificare gli RMRJ ottenuti nel passaggio precedente da BASiNET. Se il RMRJ di un circRNA è riconosciuto come RNA codificante, allora questo circRNA può essere identificato come un circRNA tradotto.
La previsione dei peptidi tradotti da RMRJs
Poiché l’espressione dei CIRCRNA negli organismi è bassa, i dati Ribo-seq non mostrano chiaramente l’esatta periodicità 3-nt nel caso di un minor numero di RPF. Pertanto, è difficile determinare l’esatto sito di inizio traduzione di un circRNA tradotto. A causa della presenza di un codone stop in alcuni RMRJ e poiché il codone start è difficile da determinare, il metodo per trovare un ORF basato su un codone start e un codone stop non è fattibile.
Per determinare le vere regioni di traduzione di questi CIRRNA e generare il prodotto di traduzione finale, FragGeneScan (Rho et al., 2010), che può predire le regioni proteina-codificanti in geni frammentati e geni con frameshifts, è usato per determinare i peptidi tradotti prodotti da circRNAs.
Per evitare l’ingombrante processo in esecuzione, tutti i modelli possono essere chiamati da uno script di shell; l’utente può semplicemente compilare il file di configurazione specificato e inserirlo nello script, e l’intero processo per prevedere i CIRCRNA tradotti verrà quindi eseguito. Inoltre, CircCode può essere eseguito separatamente, passo dopo passo, in modo tale che l’utente possa regolare i parametri nel mezzo della procedura e visualizzare i risultati di ogni passaggio come desiderato.
Risultati e discussione
Dopo il test su più computer, CircCode è stato trovato per funzionare correttamente con le dipendenze richieste installate. Per testare le prestazioni di CircCode, abbiamo utilizzato i dati per gli esseri umani e A. thaliana per prevedere CIRCRNA con potenziale di traduzione. I risultati sono stati confrontati con CIRCRNA che sono stati verificati sperimentalmente come conferma. Successivamente, abbiamo testato ulteriormente il valore False Discovery Rate (FDR) di CircCode. Abbiamo usato GenRGenS (Ponty et al., 2006) per generare un set di dati per il test basato su CIRCRNA tradotti noti e ha confermato che il valore FDR era all’interno di un intervallo accettabile e ad un livello basso. Infine, abbiamo valutato l’effetto di diverse profondità di sequenziamento dei dati Ribo-seq sulle previsioni CircCode e confrontato CircCode con altri software.
circRNAs tradotto negli esseri umani e A. thaliana
Per applicare lo strumento CircCode ai dati reali, abbiamo prima scaricato i file tra cui il genoma di riferimento umano GRCh38, genome annotation e human rRNA, da Ensembl. Per A. thaliana, i genomi di riferimento (TAIR10), i file di annotazione del genoma e le sequenze rRNA corrispondenti sono stati tutti scaricati dalle piante Ensembl. I dati Ribo-seq per gli esseri umani e A. thaliana sono stati scaricati da RPFdb (numeri di adesione: GSE96643, GSE81295, GSE88794) (Hsu et al., 2016; Willems et al., 2017), e tutti i candidati circRNAs da umano e A. thaliana sono stati scaricati da CIRCPedia v2 (Dong et al., 2018) e PlantcircBase, rispettivamente (Chu et al., 2017). In definitiva, abbiamo identificato 3.610 CIRCRNA tradotti da umani e 1.569 CIRCRNA tradotti da A. thaliana utilizzando CircCode (Dati supplementari 1).
Arricchimento funzionale di circRNAs umani e A. thaliana con potenziale di codifica
Utilizzando i risultati CircCode per umani e A. thaliana, lo strumento online KOBAS 3.0 (Wu et al., 2006) è stato impiegato per annotare questi CIRCRNA tradotti in base ai loro geni genitori. Inoltre, abbiamo eseguito l’analisi funzionale GO (Gene Ontology) e l’analisi di arricchimento KEGG (Kyoto Encyclopedia of Genes and Genomes) per questi CIRCRNA tradotti utilizzando il pacchetto R clusterProfiler (Yu et al., 2012).
I risultati di KEGG hanno mostrato che i CIRCRNA umani sono stati arricchiti nel trattamento delle proteine nella via del reticolo endoplasmatico, nella via del metabolismo del carbonio e nella via di trasporto dell’RNA. L’analisi GO ha indicato la partecipazione dei CIRCRNA tradotti umani nella regolazione del legame molecolare, dell’attività ATPasi e di altri processi biologici legati allo splicing dell’RNA. Inoltre, i CIRRNA tradotti di A. thaliana sono arricchiti in percorsi legati alla resistenza allo stress, suggerendo che svolgono un ruolo vitale in questo processo (Dati supplementari 2).
Test di Precisione per CircCode
Per indagare la precisione di CircCode, sequenze di test generati da GenRGenS, che utilizza il modello di Markov nascosto per produrre sequenze che hanno le stesse caratteristiche di sequenza (ad esempio le frequenze di nucleotidi diversi, diversi codoni e diversi nucleotidi all’inizio della sequenza), sono stati utilizzati.
Per questo studio, abbiamo usato circRNAs tradotti umani precedentemente pubblicati (Yang et al., 2017) come input per GenRGenS e generato 10.000 sequenze per testare CircCode. Abbiamo ripetuto il test 10 volte e, in media, sono stati previsti 27 circRNAs tradotti ogni volta. Il valore FDR è stato calcolato come 0,0027, che è molto inferiore a 0,05, indicando che i risultati previsti sono credibili.
Inoltre, abbiamo confrontato i CIRCRNA tradotti dagli esseri umani identificati da CircCode con i dati circRNA associati ai polisomi verificati (Yang et al., 2017). Tra questi, il 60% dei CIRCRNA è stato identificato dal CircCode (Dati supplementari 3).
Influenza della profondità di sequenziamento dei dati Ribo-seq
Per studiare l’impatto della profondità di sequenziamento dei dati Ribo-seq sui risultati di identificazione del CircCode, abbiamo prima testato l’effetto della profondità di sequenziamento sul numero di CIRCRNA tradotti (Figura 2A). Quando la profondità di sequenziamento era bassa, il numero previsto di CIRCRNA tradotte era basso e il numero di CIRCRNA tradotte aumentava con l’aumentare della profondità di sequenziamento. Il numero di CIRCRNA tradotti è diventato stabile quando la profondità di sequenziamento ha raggiunto non meno di 10× copertura trascrizione lineare.

Figura 2 (A) Effetto della profondità di sequenziamento dei dati Ribo-seq sul numero previsto di CIRCRNA tradotti. (B) L’effetto del numero di lettura della giunzione (JRN) sulla sensibilità del CircCode a diverse profondità di sequenziamento.
In secondo luogo, è stata valutata anche l’influenza di NMJ sulla sensibilità a diverse profondità di sequenziamento (Figura 2B). I risultati hanno mostrato che NMJ ha avuto un impatto minore sulla sensibilità all’aumentare della profondità di sequenziamento. CircCode aveva anche una maggiore sensibilità quando si utilizzavano dati Ribo-seq con una maggiore profondità di sequenziamento.
Confronto del CircCode con altri strumenti
Per confrontare il CircCode con altri strumenti, come CircPro, lo stesso set di dati Ribo-seq (SRR3495999) di A. thaliana è stato utilizzato per identificare i CIRCRNA tradotti utilizzando sei processori, con 16 gigabyte di RAM. CircPro ha identificato 44 CIRCRNA tradotte in 13 min, mentre CircCode ha identificato 76 CIRCRNA tradotte in 20 min. Pertanto, CircCode è più sensibile di CircPro allo stesso livello hardware del computer, ma richiede più tempo. CircPro è conciso e richiede meno tempo di CircCode, ma CircCode può identificare più circRNAs con capacità di codifica di CircPro.
Conclusioni
I CIRRNA svolgono un ruolo importante in biologia ed è fondamentale identificare con precisione i CIRRNA con capacità di codifica per la ricerca successiva. Sulla base di Python 3, abbiamo sviluppato CircCode, uno strumento da riga di comando facile da usare che ha alta sensibilità per identificare circRNAs tradotti da Ribo-Seq legge con elevata precisione. CircCode presenta buone prestazioni sia nelle piante che negli animali. Il lavoro futuro aggiungerà l’analisi dei caratteri a valle a CircCode visualizzando ogni fase del processo e ottimizzando l’accuratezza della previsione.
Disponibilità e Requisiti
CircCode è disponibile a https://github.com/PSSUN/CircCode; sistema operativo: Linux, linguaggi di programmazione: Python 3 e R; altri requisiti: bedtools (versione 2.20.0 o versione successiva), papillon, STELLE, Python 3 pacchetti (Biopython, Panda, rpy2), R-pacchetti (BASiNET, Biostrings). I pacchetti di installazione per tutto il software richiesto sono disponibili sulla homepage CircCode. Gli utenti non hanno bisogno di scaricarli singolarmente. La home page CircCode fornisce anche manuali utente dettagliati per riferimento. Lo strumento è liberamente disponibile. Non ci sono restrizioni sull’uso da parte di non accademici.
Dichiarazione di disponibilità dei dati
Tutti i dati rilevanti sono contenuti nel manoscritto e nei relativi file informativi di supporto.
Contributi dell’autore
Concettualizzazione: PS, GL. Cura dei dati: PS, GL. Analisi formale: PS, GL. Scrittura-Bozza originale: PS, GL. Scrittura-Revisione e modifica: PS, GL.
Finanziamento
Questo lavoro è stato sostenuto dalle sovvenzioni della National Natural Science Foundation of China (grant nn. 31770333, 31370329 e 11631012), dal Programma per i talenti eccellenti del Nuovo secolo nell’università (NCET-12-0896) e dai fondi di ricerca fondamentali per le Università centrali (n. GK201403004). Le agenzie di finanziamento non hanno avuto alcun ruolo nello studio, nella sua progettazione, nella raccolta e nell’analisi dei dati, nella decisione di pubblicare o nella preparazione del manoscritto. I finanziatori non avevano alcun ruolo nella progettazione dello studio, nella raccolta e nell’analisi dei dati, nella decisione di pubblicare o nella preparazione del manoscritto.
Conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di rapporti commerciali o finanziari che potrebbero essere interpretati come un potenziale conflitto di interessi.
Materiale supplementare
Il materiale supplementare per questo articolo può essere trovato online all’indirizzo: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
Dati supplementari 1 / La sequenza del circRNA tradotto predetto e peptide corto.
Dati supplementari 2 / Risultati di arricchimento di GO e di arricchimento di KEGG per l’uomo e Arabidopsis thaliana.
Dati supplementari 3 / Confronto tra CIRCRNA tradotti previsti e CIRCRNA tradotti convalidati.
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: un trimmer flessibile per i dati di sequenza illumina. Bioinformatica 30, 2114-2120. doi: 10.1093 / bioinformatica / btu170
PubMed Abstract / CrossRef Full Text / Google Scholar
Brar, G. A., Weissman, J. S. (2015). Il profilo del ribosoma rivela cosa, quando, dove e come della sintesi proteica. NAT. Rev. Mol. Biol cellulare. 16, 651–664. doi: 10.1038 | nrm4069
PubMed Abstract / CrossRef Full Text / Google Scholar
Chen, L.-L., Yang, L. (2015). Regolazione della biogenesi circRNA. RNA Biol. 12, 381–388. doi: 10.1080/15476286.2015.1020271
PubMed Abstract / CrossRef Testo completo / Google Scholar
Chu, Q., Zhang, X., Zhu, X., Liu, C., Mao, L., Ye, C., et al. (2017). PlantcircBase: un database per gli RNA circolari delle piante. Mol. Pianta 10, 1126-1128. doi: 10.1016 / j. molp.2017.03.003
PubMed Abstract | CrossRef Testo completo / Google Scholar
Dobin, A., Davis, C. A., Schlesinger, F.,Drenkow, J., Zaleski, C., Gai, S., et al. (2013). STELLA: allineamento RNA-seq universale ultraveloce. Bioinformatica 29, 15-21. doi: 10.1093 / bioinformatica / bts635
PubMed Abstract / CrossRef Full Text / Google Scholar
Dong, R., Ma, X.-K., Chen, L.-L., Yang, L. (2019). “Genome-wide annotation of circRNAs and their alternative back-splicing / splicing with CIRCexplorer Pipeline,” in Epitranscriptomics. Eds. Il suo nome deriva dal latino “”Wajapeyee, N., Gupta, R. “” (New York, NY: Springer New York), 137-149. doi: 10.1007/978-1-4939-8808-2_10
CrossRef Testo completo / Google Scholar
Dong, R., Ma, X.-K., Li, G.-W., Yang, L. (2018). CIRCpedia v2: un database aggiornato per l’annotazione circolare completa dell’RNA e il confronto delle espressioni. Genomica Proteomica Bioinf. 16, 226–233. doi: 10.1016 / j. gpb.2018.08.001
CrossRef Testo completo / Google Scholar
Gao, Y., Wang, J., Zhao, F. (2015). CIRI: un algoritmo efficiente e imparziale per l’identificazione circolare di RNA de novo. Genome Biol. 16, 4. doi: 10.1186 / s13059-014-0571-3
PubMed Abstract / CrossRef Testo completo / Google Scholar
Nel 2013 è stato pubblicato il primo album in studio del gruppo musicale statunitense Guttman, pubblicato nel 2013. Il profilo del ribosoma fornisce la prova che i grandi RNA non codificanti non codificano le proteine. Cella 154, 240-251. doi: 10.1016 / j.cella.2013.06.009
PubMed Abstract | CrossRef Testo completo / Google Scholar
Hsu, P. Y., Benfey, P. N. (2018). Piccolo ma potente: peptidi funzionali codificati da piccoli ORFs nelle piante. PROTEOMICA 18, 1700038. doi: 10.1002 / pmic.201700038
CrossRef Testo completo / Google Scholar
Il suo nome deriva dal latino “Hsu”, “P. Y.”, “Calviello”, “L.”, “Wu”, “H.-Y. L.”, “Li”, “F.-W.”, “Rothfels”, “C. J.”, “Ohler”, “U.”, ” et al. (2016). La profilazione del ribosoma a super risoluzione rivela eventi di traduzione non annotati in Arabidopsis. Proc. Natl. Acad. Sic. 113, E7126–E7135. doi: 10.1073 / pnas.1614788113
CrossRef Testo completo / Google Scholar
Ingolia, NT, Lareau, LF, Weissman, JS (2011). Il profilo del ribosoma delle cellule staminali embrionali del topo rivela la complessità e la dinamica dei proteomi dei mammiferi. Cella 147, 789-802. doi: 10.1016 / j.cella.2011.10.002
PubMed Abstract | CrossRef Testo completo / Google Scholar
It, E. A., Katahira, I., Vicente, F. F., da, R., Pereira, L. F. P., Lopes, F. M. (2018). BASiNET-Biological Sequences NETwork: un caso di studio sull’identificazione di RNA codificanti e non codificanti. Acidi nucleici Res. 46, e96–e96. doi: 10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093 / bioinformatica / bty948
CrossRef Testo completo / Google Scholar
Kearse, MG, Wilusz, J. E. (2017). Traduzione non AUG: un nuovo inizio per la sintesi proteica negli eucarioti. Geni Dev. 31, 1717–1731. doi: 10.1101 / gad.305250.117
PubMed Abstract | CrossRef Testo completo / Google Scholar
La maggior parte di essi sono di origine cinese. (2007). CPC: valutare il potenziale di codifica delle proteine dei trascritti utilizzando le funzioni di sequenza e la macchina vettoriale di supporto. Acidi nucleici Res. 35, W345–W349. doi: 10.1093 / nar / gkm391
PubMed Abstract / CrossRef Testo completo / Google Scholar
Nel 2012 si trasferisce in Germania. Veloce gapped-leggere l’allineamento con Bowtie 2. NAT. Metodi 9, 357-359. doi: 10.1038 / nmeth.1923
PubMed Abstract / CrossRef Testo completo / Google Scholar
Meng, X., Chen, Q., Zhang, P., Chen, M. (2017). CircPro: uno strumento integrato per l’identificazione di CIRCRNA con potenziale di codifica delle proteine. Bioinformatica 33, 3314-3316. doi: 10.1093 / bioinformatica/btx446
PubMed Abstract / CrossRef Testo completo / Google Scholar
Michel, A. M., Baranov, P. V. (2013). Ribosome profiling: un monitor Hi-Def per la sintesi proteica su scala genome-wide: ribosome profiling. Wiley Interdiscip. Rev. RNA 4, 473-490. doi: 10.1002 / wrna.1172
PubMed Abstract / CrossRef Testo completo / Google Scholar
Pamudurti, N. R., Bartok, O., Jens, M., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., et al. (2017). Traduzione di CircRNAs. Mol. Cella 66, 9-21.e7. doi: 10.1016 / j.molcel.2017.02.021
PubMed Abstract / CrossRef Testo completo / Google Scholar
Ponty, Y., Termier, M., Denise, A. (2006). GenRGenS: software per la generazione di sequenze e strutture genomiche casuali. Bioinformatica 22, 1534-1535. doi: 10.1093 / bioinformatica / btl113
PubMed Abstract / CrossRef Full Text / Google Scholar
Rho, M., Tang, H., Ye, Y. (2010). FragGeneScan: predire i geni in letture brevi e soggette a errori. Acidi nucleici Res. 38, e191–e191. doi: 10.1093/nar/gkq747
PubMed Astratto | CrossRef Testo Completo | Google Scholar
Slavoff, S. A. Mitchell, A. J., Schwaid, A. G., di Cabili, M. N., Ma, J., Levin, J. Z., et al. (2013). Scoperta peptidomica di peptidi con codifica frame a lettura aperta in cellule umane. NAT. Chimica. Biol. 9, 59–64. doi: 10.1038 / nchembio.1120
PubMed Abstract / CrossRef Testo completo / Google Scholar
Spealman, P., Naik, A. W., Maggio, G. E., Kuersten, S., Freeberg, L., Murphy, RF, et al. (2018). uORFs non AUG conservati rivelati da una nuova analisi di regressione dei dati di profilazione dei ribosomi. Genoma Res. 28, 214-222. doi: 10.1101 / gr.221507.117
PubMed Abstract | CrossRef Testo completo / Google Scholar
Vicens, Q., Westhof ,E. (2014). Biogenesi degli RNA circolari. Cella 159, 13-14. doi: 10.1016 / j.cella.2014.09.005
PubMed Abstract | CrossRef Testo completo / Google Scholar
Willems, P., Ndah, E., Jonckheere, V., Stael, S., Sticker, A., Martens, L., et al. (2017). La proteomica N-terminale ha assistito la profilazione del paesaggio di iniziazione della traduzione inesplorato in Arabidopsis thaliana. Mol. Cellula. Proteomica 16, 1064-1080. doi: 10.1074 / mcp.M116. 066662
PubMed Abstract | CrossRef Testo completo / Google Scholar
Wu, J., Mao, X., Cai, T., Luo, J., Wei, L. (2006). KOBAS server: una piattaforma web per l’annotazione automatizzata e l’identificazione del percorso. Acidi nucleici Res. 34, W720–W724. doi: 10.1093 / nar / gkl167
PubMed Abstract / CrossRef Full Text / Google Scholar
Yang, L., Fu, J., Zhou, Y. (2018). RNA circolari e loro ruoli emergenti nella regolazione immunitaria. Anteriore. Immunolo. 9, 2977. doi: 10.3389 / fimmu.2018.02977
PubMed Abstract | CrossRef Testo completo / Google Scholar
Yang, Y., Fan, X., Mao, M., Song, X., Wu, P., Zhang, Y., et al. (2017). Ampia traduzione di RNA circolari guidati da N6-metiladenosina. Cella 27, 626-641. doi: 10.1038 / cr.2017.31
PubMed Abstract / CrossRef Testo completo / Google Scholar
Nel 2012 è stato pubblicato il primo album in studio. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar