- Introduzione
- ChREBP Struttura e regolazione tramite i domini LID/Grace
- Attivazione di ChREBP da parte dei metaboliti del glucosio
- Cofattori e partner di ChREBP
- Ruolo di ChREBP nel metabolismo dei carboidrati e nella produzione di epatochine
- ChREBP come regolatore della sintesi degli acidi grassi epatici e della secrezione di VLDL
- Regolazione del metabolismo del fruttosio da parte di ChREBP nel fegato e nell’intestino
- Regolamento del BDK:Asse PPM1K nel fegato
- ChREBP è necessario per la regolazione mediata dal glucosio di FGF21
- Ruolo di ChREBP in Inter-Organo di Rete Che Controlla l’Omeostasi Energetica
- Funzione Epatica ChREBP nel Controllo della Sensibilità all’Insulina Equilibrio
- Il ChREBP adiposo collega la lipogenesi alla sensibilità all’insulina
- Nuova interazione tra lipasi sensibile all’ormone e ChREBP nel tessuto adiposo
- Conclusione e direzioni future
- Contributi degli autori
- Finanziamento
- Dichiarazione sul conflitto di interessi
Introduzione
Aumento del consumo di zuccheri semplici come il saccarosio e ad alto contenuto di fruttosio sciroppo di mais negli ultimi anni ha portato ad un aumento del rischio di malattie metaboliche come l’obesità, la dislipidemia, il diabete di tipo 2 e/o steatosi epatica non alcolica (NAFLD). Il fegato è l’organo principale responsabile della conversione dei carboidrati alimentari in eccesso in grassi. I trigliceridi risultanti (TG) possono essere confezionati in lipoproteine a densità molto bassa (VLDL) e secreti nella circolazione, immagazzinati come goccioline lipidiche o metabolizzati attraverso la via di beta-ossidazione. L’insulina secreta in risposta a glicemia elevata, stimola l’espressione dei geni della sintesi degli acidi grassi de novo (lipogenesi) attraverso il fattore di trascrizione sterol regulatory element binding protein-1c (SREBP-1c) (Foretz et al., 1999). SREBP-1c agisce in sinergia con un altro fattore di trascrizione chiamato proteina legante dell’elemento di risposta del carboidrato (ChREBP), che media la risposta ai carboidrati dietetici. La struttura della proteina ChREBP contiene un dominio inibitorio del glucosio basso (LID) e un elemento conservato di attivazione della glucosio-risposta (GRACE) situato nel suo N-terminale (Li et al., 2006). L’attivazione del dominio di GRACE da parte dei metaboliti del glucosio promuove l’attività trascrizionale di ChREBP e il legame con una sequenza altamente conservata chiamata elemento di risposta dei carboidrati (ChoRE). ChoRE è presente sui promotori dei geni target ChREBP, che codificano gli enzimi chiave della lipogenesi de novo tra cui la L-piruvato chinasi (L-pk), un enzima limitante la velocità nella glicolisi, la sintasi degli acidi grassi (Fas), l’acetil-CoA carbossilasi (Acc) e la stearoil-CoA desaturasi (Scd1) (Kawaguchi et al., 2001). Un recente studio ha riportato l’interdipendenza tra ChREBP (attivato dal glucosio) e SREBP-1c (attivato dall’insulina) per la piena induzione dell’espressione genica glicolitica e lipogenica nel fegato (Linden et al., 2018). Il ristabilimento virale della forma attiva nucleare di SREBP-1c nel fegato dei topi ChREBP-carenti (ChREBPKO) ha normalizzato l’espressione genica lipogenica, pur non avendo alcun effetto sul salvataggio dell’espressione genica glicolitica. L’esperimento mirror, in cui l’espressione di ChREBP è stata indotta nel fegato di topi knockout SREBP-1c, ha salvato l’espressione genica glicolitica ma sorprendentemente non l’espressione genica lipogenica, nonostante il ben noto ruolo di ChREBP nel controllo dei geni di sintesi degli acidi grassi. Tuttavia, questo studio suggerisce l’importanza della doppia azione di ChREBP e SREBP-1c nei geni di controllo coinvolti nella regolazione della sintesi degli acidi grassi (Linden et al., 2018).
ChREBP è altamente arricchito nel fegato ed è stato studiato come regolatore principale del metabolismo lipidico (Iizuka et al., 2004; Osorio et al., 2016). Il ChREBP è anche significativamente espresso nelle isole pancreatiche, nell’intestino tenue, nel muscolo scheletrico e in misura minore nel rene e nel cervello (vedi Richards et al., 2017 per la revisione). È interessante notare che un’altra isoforma di ChREBP, ChREBPß, proveniente da un primo promotore di esone alternativo, è stata identificata per la prima volta nel tessuto adiposo (Herman et al., 2012) e successivamente descritto in altri tipi di cellule (vedi Abdul – Wahed et al., 2017 per la revisione). Come discuteremo, ChREBPß è descritto come un’isoforma costitutivamente attiva. Si spera che il lavoro futuro affronterà i rispettivi ruoli di ChREBP e ChREBPß nella regolazione del metabolismo del glucosio e dei lipidi e identificherà i loro obiettivi specifici e/o sovrapposti.
ChREBP Struttura e regolazione tramite i domini LID/Grace
ChREBP appartiene alla famiglia Mondo dei fattori di trascrizione bHLH/Zip. Il dominio N-terminale (residui 1-251) contiene due segnali di esportazione nucleare (NES) e un segnale di localizzazione nucleare (NLS) che regola la localizzazione subcellulare interagendo con il mantenimento della regione cromosomica 1 (CRM1) noto anche come proteine exportin 1 e/o 14-3-3 (Sakiyama et al., 2008). La regione C-terminus contiene un dominio poliprolina, un dominio bHLH/LZ (660-737 residui) e un dominio leucina zipper-like (Zip-like, 807-847 residui) che sono associati a co-fattori e legame al DNA (Yamashita et al., 2001; Fukasawa et al., 2010; Ge et al., 2012). La localizzazione e l’attivazione trascrizionale di ChREBP sono determinate dalla disponibilità di nutrienti. La regolazione mediata dal glucosio di ChREBP avviene principalmente a livello del modulo di rilevamento del glucosio (GSM) o della regione mondo conservata( MCR), che è composto dal LID e dai domini di GRAZIA, come menzionato nell’introduzione (Figura 1A; Li et al., 2006; Singh e Irwin, 2016). Nel 2012, Herman et al. (2012) ha descritto un’altra isoforma di ChREBP, ChREBPß, che viene trascritta da un primo promotore di esone alternativo 1b all’esone 2 (Figura 1B). Questa trascrizione è tradotta dall’esone 4, generando una proteina più corta di 687 aminoacidi (l’isoforma ChREBP a tutta lunghezza, ribattezzata α, contiene 864 aminoacidi, chiamati ChREBP nel manoscritto) in cui mancano i due NES, NLS e il dominio LID. ChREBPß è altamente attivo nel tessuto adiposo bianco in modo dipendente da GLUT-4 e si suggerisce di essere direttamente regolato da ChREBPa poiché una sequenza di lavoro è stata identificata nel promotore dell’esone 1β (Herman et al., 2012; Figura 1B). La regolazione di ChREBPß da parte di ChREBPa suggerisce l’esistenza di un ciclo feed-forward che potenzialmente aggrava la risposta al glucosio in condizioni iperglicemiche. Tuttavia, è necessario chiarire i meccanismi di regolamentazione dell’isoforma ChREBPß e, cosa più importante, la sua funzione specifica.
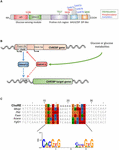
Figura 1. (A) Struttura dell’elemento di risposta dei carboidrati che lega la proteina α (ChREBPa). ChREBPa è composto da 864 aminoacidi e contiene diversi domini regolatori. All’estremità N la proteina contiene un modulo di rilevamento del glucosio composto dal dominio inibitorio del glucosio basso (LID) e dall’elemento conservato attivato dal glucosio (GRACE). La proteina contiene anche un ricco di poliprolina, un bHLH / LZ e un dominio simile alla leucina (Zip-like) situato al C-terminale. Le modifiche post-traduzionali sono indicate nei rispettivi residui, nella fosforilazione (rosso), nell’acetilazione (blu) e nelle O-GlcNAcilazioni recentemente identificate (verde). B) Struttura genica del gene ChREBP e generazione delle due isoforme ChREBP α e β. ChREBPß è trascritto da un promotore di primo esone alternativo 1b. Questa trascrizione è tradotta dall’esone 4 generando una proteina più corta di 687 aminoacidi in cui mancano i due NES, l’NLS e il dominio LID. L’isoforma di ChREBPß è stata suggerita per essere regolata direttamente da ChREBPa poiché una sequenza di lavoro è stata identificata nel promotore dell’esone 1b. Se entrambe le isoforme di ChREBP α e β si legano entrambe al lavoro non è attualmente noto. Figura adattata da Herman et al. (2012). (C) Il multi-allineamento delle sequenze di consenso di lavoro di routine presenta in parecchi promotori del gene dell’obiettivo di ChREBP. L’allineamento basato sui nucleotidi è presentato nella parte superiore della figura insieme al lavoro di routine della sequenza di consenso descritto in Poungvarin et al. (2015). È rappresentato anche il logo corrispondente alla sequenza di consenso associata a questo particolare allineamento.
Attivazione di ChREBP da parte dei metaboliti del glucosio
In condizioni di digiuno, l’attivazione glucagone-dipendente della protein chinasi A (PKA) (Kawaguchi et al., 2002) fosforila ChREBP sui residui Ser196 e Thr666, portando al legame di ChREBP alla proteina 14-3-3 e alla sua ritenzione nel citosol (Kawaguchi et al., 2001, 2002; Davies et al., 2008). AMP activated protein chinase (AMPK), un sensore di energia cellulare centrale, fosforila anche ChREBP sul residuo Ser568, che a sua volta diminuisce il legame di ChREBP ai promotori dei suoi geni bersaglio (Kawaguchi et al., 2002; Sato et al., 2016). È stato dimostrato che i metaboliti generati durante il digiuno, come i corpi AMP e chetonici prodotti dall’ossidazione degli acidi grassi, svolgono un ruolo inibitorio allosterico alterando l’affinità proteica ChREBP e 14-3-3, migliorando la stabilizzazione complessa e favorendo la ritenzione citosolica (Sakiyama et al., 2008; Nakagawa et al., 2013; Sato et al., 2016). In risposta ai carboidrati, il ChREBP è regolato a livello trascrizionale, traslazionale e post-traslazionale. L’aumento delle concentrazioni di glucosio dopo un pasto promuove la sintesi di metaboliti intermedi come xilulosio-5-fosfato (X5P), inizialmente proposto come attivatore della proteina fosfatasi 2A (PP2A) (Kawaguchi et al., 2001; Kabashima et al., 2003). PP2A è stato precedentemente descritto per defosforilare ChREBP al residuo Ser196 permettendo la sua traslocazione al nucleo dove è ulteriormente defosforilato in modo X5P-e PP2A-dipendente (su Thr666 e Ser568). Tuttavia, questo modello è stato sfidato nel corso degli anni e altri metaboliti come il glucosio-6-fosfato (G6P) sono stati proposti come potenziali attivatori della traslocazione/attività di ChREBP (Dentina et al., 2012). McFerrin et al. (2012) ha identificato un motivo putativo per il legame G6P (253-SDTLFT-258) sul dominio GRACE, che è anche conservato in MondoA, un ortholog ChREBP/MondoB (vedi Richards et al., 2017 per la revisione). Secondo questa ipotesi, G6P potrebbe promuovere un cambiamento di conformazione allosterica che induce una conformazione aperta per ChREBP, facilitando l’interazione con i cofattori e la successiva traslocazione al nucleo (McFerrin et al., 2012).
All’interno del nucleo, il ChREBP può essere modificato attraverso la O-glcnacilazione, una modifica post-traslazionale dipendente dal metabolismo del glucosio e identificata come importante per l’attività trascrizionale del ChREBP (Guinez et al., 2010). La O-glcnacilazione si verifica sui residui di serina e treonina attraverso l’attività della O-GlcNAc transferasi (OGT), un enzima che aggiunge residui di N-acetilglucosamina (GlcNAc) alle proteine bersaglio modificando così la loro attività, stabilità e/o posizione subcellulare. Yang et al. (2017) ha recentemente rivelato diversi residui di ChREBP modificati dalla O-glcnacilazione. Mutazioni di questi residui nei domini bHLH/ZIP e dimerizzazione e dominio di localizzazione citoplasmatica (DCD) hanno permesso l’identificazione di Thr517 e Ser839 come siti essenziali per l’attivazione glucosio-dipendente di ChREBP (Figura 1A). ChREBP può anche essere modificato mediante acetilazione tramite l’attività dell’istone acetiltransferasi di p300 (Bricambert et al., 2010). p300 attivato dal glucosio acetila ChREBP su Lys672 e aumenta la sua attività trascrizionale migliorando il suo reclutamento nella sequenza di lavoro, che è la sequenza di legame ottimale del consenso è CAYGYCnnnnnCRCRTG (Figura 1C). Poungvarin et al. (2015) analizzato siti di legame ChREBP da ChIP-seq nel fegato e nel tessuto adiposo bianco di topi ri-alimentati con una dieta ricca di carboidrati e senza grassi. Hanno riferito che il legame di ChREBP è arricchito in vie coinvolte nella segnalazione dell’insulina, nelle giunzioni aderenti e nel cancro, suggerendo un nuovo coinvolgimento di ChREBP nella tumorigenesi e nella progressione del cancro. Inoltre, un recente studio ha riportato l’importanza di ChREBP nel carcinoma epatocellulare (HCC) (Ribback et al., 2017). Gli autori hanno scoperto che la delezione genetica di ChREBP (nei topi ChREBPKO) ha compromesso l’epatocarcinogenesi guidata dalla sovraespressione della proteina chinasi B/Akt nei topi. Inoltre, l’inibizione di ChREBP mediata da siRNA in cellule HCC di topo e / o umane ha determinato una diminuzione della proliferazione e dell’apoptosi.
Cofattori e partner di ChREBP
Diversi cofattori e/o partner di ChREBP sono stati identificati negli ultimi anni (vedi Richards et al., 2017 per la revisione). Max like protein x (Mlx), un fattore di trascrizione bHLH/LZ, è stato il primo identificato come partner di legame comune della famiglia Mondo (Stoeckman et al., 2004). La dimerizzazione di ChREBP con Mlx è richiesta sia per la traslocazione nucleare in risposta al glucosio che per il legame con gli elementi di routine. I recettori nucleari come il fattore nucleare degli epatociti 4α (HNF4a) e il recettore X farnesoide (FXR) sono stati anche descritti come partner di ChREBP. HNF4a interagisce fisicamente con ChREBP legandosi alla regione diretta repeat-1 (DR-1) sul promotore dei geni target ChREBP (Adamson et al., 2006; Meng et al., 2016). Inoltre, le proteine del co-attivatore trascrizionale p300 / CBP hanno dimostrato di stabilizzare il complesso ChREBP / HNF4a (Burke et al., 2009). Le proteine del co-attivatore trascrizionale p300 / CBP svolgono un ruolo centrale nel coordinare e integrare più eventi dipendenti dal segnale con l’apparato di trascrizione. Un’altra proprietà chiave di p300 / CBP è la presenza dell’attività dell’istone acetiltransferasi (HAT), che conferisce a p300/CBP la capacità di influenzare l’attività della cromatina modulando gli istoni nucleosomiali. Negli epatociti umani, il legame FXR con il complesso ChREBP-HNF4a innesca il rilascio di ChREBP da CBP / p300, portando al reclutamento dell’istone deacetilasi SMRT sul promotore Lpk, agendo così come co-repressore dell’attività trascrizionale di ChREBP (Caron et al., 2013). Inoltre, l’attività CBP / p300 HAT modifica ChREBP su Lys 672, portando alla sua attivazione trascrizionale in risposta al glucosio (Bricambert et al., 2010).
Bricambert et al. (2018) ha recentemente identificato l’istone demethylase plant homeodomain finger 2 (Phf2), che appartiene alla famiglia dell’istone lisina demetilasi (KDM7), come un nuovo co-fattore di ChREBP. L’interazione tra Phf2 e ChREBP migliora l’attivazione trascrizionale di ChREBP cancellando i segni di metile H3K9 sul promotore dei suoi geni bersaglio. È interessante notare che il co-reclutamento specifico di Phf2 e ChREBP al promotore del fattore nucleare eritroide 2 come 2 (Nrf2) contribuisce all’effetto protettivo di Phf2 contro l’aumento delle specie reattive dell’ossigeno (ROS) e la progressione della NAFLD nel contesto dell’iperglicemia (Bricambert et al., 2018).
Ruolo di ChREBP nel metabolismo dei carboidrati e nella produzione di epatochine
ChREBP come regolatore della sintesi degli acidi grassi epatici e della secrezione di VLDL
La malattia del fegato grasso non alcolica è un segno distintivo della sindrome metabolica e gli studi sugli esseri umani rivelano che la lipogenesi de novo contribuisce a circa il 25% dei lipidi, 2005). Negli stati insulino-resistenti, l’iperglicemia e l’iperinsulinemia aumentano la lipogenesi in parte attraverso l’attivazione di ChREBP e SREBP-1c. Inibizione ChREBP nel fegato di topi obesi e insulino-resistenti ob/ob, attraverso RNAi o ablazione genetica porta all’inversione della steatosi epatica (Dentina et al., 2006; Iizuka et al., 2006). La secrezione alterata di VLDL dal fegato contribuisce anche alla patogenesi della NAFLD. La proteina microsomiale di trasferimento dei trigliceridi (MTTP)è la proteina responsabile dell’assemblaggio e della secrezione delle lipoproteine contenenti apolipoproteina B. La carenza di MTTP nei topi e nell’uomo causa ipolipidemia e fegato grasso. La regolazione di questa proteina è stata associata con alcuni cis-elementi altamente conservati nel suo promotore compreso i domini regolatori positivi e negativi critici (Cuchel et al., 2013; Hussain et al., 2011). Recentemente, ChREBP è stato indicato come un potenziale regolatore di MTTP poiché la mancanza di ChREBP funzionale nel fegato sopprime l’espressione di Mttp e l’assemblaggio e la secrezione di VLDL (Niwa et al., 2018). Tuttavia, poiché non è stato possibile identificare chiaramente alcun compito sul promotore dell’MTTP, saranno necessarie ulteriori analisi per identificare il meccanismo con cui la ChREBP regola l’MTTP.
Regolazione del metabolismo del fruttosio da parte di ChREBP nel fegato e nell’intestino
Il legame tra ChREBP e il metabolismo del fruttosio è stato evidenziato per la prima volta dall’analisi fenotipica dei topi knockout di ChREBP (topi ChREBPKO). I topi ChREBPKO sono stati segnalati per morire entro diversi giorni di alimentazione ad alto contenuto di fruttosio (HFrD) (Iizuka et al., 2004). Questa importante intolleranza al fruttosio è stata attribuita alla riduzione dell’espressione della fruttochinasi e della triosio chinasi, due enzimi necessari per il metabolismo del fruttosio (Iizuka et al., 2004). Kim et al. (2016) in seguito ha riportato l’importanza di ChREBP per l’efficiente conversione del fruttosio in glucosio nel fegato e nella clearance del fruttosio di tutto il corpo, ma anche, sotto ingestione di fruttosio, ChREBP potrebbe contribuire all’iperglicemia attivando direttamente l’espressione G6pc, un gene chiave della gluconeogenesi. Questo effetto potrebbe portare a un circolo vizioso in cui il consumo di fruttosio esacerba la produzione di glucosio attraverso l’attività di ChREBP (Kim et al., 2016). L’anno successivo, lo studio di Zhang et al. (2017) ha riferito che i topi ChREBPKO alimentati con HFrD sviluppano gravi lesioni epatiche a causa dell’eccessiva attivazione dello stress del reticolo endoplasmatico e dell’apoptosi epatocitaria mediata da proteina omologa CCAAT-enhancer-binding protein (CHOP). L’apoptosi negli epatociti in questi topi è stata probabilmente legata all’aumento della biosintesi del colesterolo poiché l’inibizione di questa via tramite HMG-CoA reduttasi (HMGCR) o l’inibizione di SREBP2 ha salvato i topi ChREBPKO dalla lesione epatica indotta da HFrD. Una mancanza di ChREBP è stata anche recentemente associata a una disregolazione del metabolismo del saccarosio e del fruttosio che porta all’intolleranza allo zucchero e al malassorbimento nei topi (Kato et al., 2018). Questi effetti sono stati associati a una diminuzione dell’espressione di saccarosio-isomaltasi intestinale (SI), che digerisce il saccarosio in glucosio e fruttosio, i trasportatori di glucosio 5 (Glut5) e 2 (Glut2) e l’enzima chetoesochinasi (Khk), che regola la fruttolisi (Figura 2). La disregolazione di questi enzimi può condurre all’accumulazione del saccarosio e del fruttosio non digeriti con le ripercussioni potenziali nella composizione del microbiota dell’intestino. Il confronto tra ChREBPKO e topi knockout CHREBP specifici per il fegato (ChREBPLiverKO) alimentati con HFrD aveva precedentemente rivelato che la carenza epatica di ChREBP da sola non porta a intolleranza al fruttosio, ma che la carenza di ChREBP nell’intestino tenue è probabilmente responsabile della compromissione della tolleranza al fruttosio osservata in questi topi (Kim et al., 2017). Complessivamente, questi studi sottolineano l’importanza di ChREBP nella regolazione del metabolismo del fruttosio e sottolineano la necessità di una migliore comprensione del suo ruolo e regolazione nell’intestino tenue.
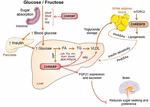
Figura 2. ChREBP regola le vie multiple di segnalazione/metaboliche in risposta a glucosio e fruttosio. ChREBP è espresso in diversi tessuti tra cui intestino, fegato e tessuto adiposo bianco. In questi tipi di cellule, in risposta al glucosio e/o fruttosio ChREBP viene attivato e induce specifico programma genico come indicato in figura. Nell’intestino, la stimolazione dell’espressione di SI, Glut5, Glut2 e ketoesochinasi (Khk) da parte di ChREBP (direttamente o indirettamente) è stata descritta per migliorare la tolleranza al saccarosio e l’assorbimento del fruttosio. Nel fegato, ChREBP è un modulatore chiave dell’espressione genica glicolitica, lipogenica e microsomiale della proteina di trasferimento dei trigliceridi (Mttp), controllando così sia l’accumulo di acidi grassi che l’esportazione di VLDL dal fegato. ChREBP è anche regola la produzione di epatochine come il fattore di crescita dei fibroblasti 21 (FGF21). Questo asse fegato-cervello espande la funzione di ChREBP del fegato da un regolatore epatico a un modulatore sistemico che influenza non solo la gestione del substrato nel fegato ma anche la preferenza dei nutrienti. L’attivazione di ChREBP nel tessuto adiposo bianco è collegata ad omeostasi metabolica migliore producendo i segnali circolanti protettivi. Una nuova classe di lipidi di mammiferi caratterizzati da un legame estere ramificato tra un acido grasso e un acido idrossi-grasso (acido palmitico idrossil stearico) è stata segnalata per esercitare effetti benefici sull’omeostasi del glucosio attraverso la modulazione diretta e incretina-mediata della funzione delle cellule β, aumento dell’assorbimento del glucosio adiposo e riduzione dell’infiammazione. È interessante notare che mTORC2 è stato recentemente identificato come un nuovo regolatore dell’isoforma di ChREBPß nelle cellule adipose.
Regolamento del BDK:Asse PPM1K nel fegato
Il primo passo impegnato del catabolismo dell’amminoacido a catena ramificata (BCAA) è regolato dal complesso chetoacido deidrogenasi a catena ramificata (BCKDH) che è controllato da due enzimi, l’alfa-chetoacido a catena ramificata deidrogenasi chinasi (BDK) e la fosfatasi proteica, Mg2+/Mn22+ 1K dipendente (PPM1K). White et al. (2018) recentemente ha associato ChREBP all’upregulation di BDK e alla down-regulation di PPM1K nel fegato e ha identificato un motivo di lavoro conservato nel promotore di entrambi questi geni. Una correlazione positiva tra l’espressione di BDK e altri geni bersaglio tipici di ChREBP (Fasn, Pklr, ChREBPß) è stata osservata nei fegati di ratti alimentati con una dieta ricca di glucosio o fruttosio. A livello fisiologico, l’aumento del rapporto BDK: PPM1K ha portato alla fosforilazione e all’attivazione della liasi ATP-citrato (ACLY), stimolando così la lipogenesi de novo. Questi risultati rivelano che BDK e PPM1K possono essere nuovi geni che attivano la lipogenesi regolati da ChREBPß. Dato il loro ruolo nella regolazione del metabolismo dei lipidi, del glucosio e degli aminoacidi, BDK e PPM1K potrebbero essere considerati potenziali bersagli terapeutici nel fegato nel prossimo futuro (White et al., 2018).
ChREBP è necessario per la regolazione mediata dal glucosio di FGF21
ChREBP è stato recentemente associato alla produzione e alla secrezione di epatochine come il fattore di crescita dei fibroblasti 21 (FGF21) (Iizuka et al., 2009; Dushay et al., 2015, Iroz et al., 2017). FGF21 è un ormone metabolico sintetizzato dal fegato con molteplici effetti benefici nei tessuti periferici (Kharitonenkov et al., 2005; Badman et al., 2007; Markan et al., 2014). Fino a poco tempo fa, FGF21 era considerato un ormone a digiuno che migliora l’ossidazione degli acidi grassi, la chetogenesi e la lipolisi sotto il controllo trascrizionale del recettore α attivato dal proliferatore del perossisoma (PPARa) (Inagaki et al., 2007). Un lavoro di routine sul promotore Fgf21 è stato precedentemente identificato in entrambi i topi (da -74 a -52 bp) e gli esseri umani (da -380 a -366 bp) (Iizuka et al., 2009) ma gli studi funzionali sono mancati fino a poco tempo fa. È stato riportato che il consumo di glucosio e fruttosio ha portato a un rapido aumento dei livelli di FGF21 in volontari sani e pazienti con sindrome metabolica (Dushay et al., 2015). Ulteriori studi hanno anche riportato un legame meccanicistico tra FGF21 derivato da ChREBP e preferenza macronutriente attraverso un asse fegato-cervello (Talukdar et al., 2016; von Holstein-Rathlou et al., 2016). Questo asse fegato-cervello espande la funzione ChREBP da un regolatore metabolico epatico a un modulatore sistemico, influenzando non solo la gestione del substrato epatico ma anche il comportamento alimentare globale (Abdul-Wahed et al., 2017).
Ruolo di ChREBP in Inter-Organo di Rete Che Controlla l’Omeostasi Energetica
Funzione Epatica ChREBP nel Controllo della Sensibilità all’Insulina Equilibrio
il Nostro laboratorio segnalato in precedenza che ChREBP agisce come una chiave modulatore epatica e composizione in acidi grassi e la sensibilità all’insulina nel contesto di bevande alcoliche e non alcoliche, malattie del fegato (vedere Abdul Wahed et al., 2017 per la revisione). I topi che sovraesprimevano ChREBP hanno sviluppato una maggiore steatosi epatica rispetto ai controlli, ma è interessante notare che sono rimasti privi di complicanze metaboliche e non hanno sviluppato insulino-resistenza. L’analisi lipidomica ha rivelato che la steatosi mediata da ChREBP è associata ad una diminuzione degli acidi grassi saturi e ad un aumento degli acidi grassi monoinsaturi, questi ultimi che hanno dimostrato di essere associati ad effetti benefici mediati da ChREBP sulla sensibilità all’insulina (Benhamed et al., 2012). Questi risultati dimostrano il ruolo del ChREBP nel partizionamento lipidico e suggeriscono che specifiche specie lipidiche, quando presenti nella posizione e nel tempo corretti, possono innescare segnali che modulano l’adattamento allo stress metabolico (Benhamed et al., 2012; Bricambert et al., 2018). È interessante notare che Jois et al. (2017) ha anche suggerito un ruolo protettivo per il ChREBP epatico per quanto riguarda l’omeostasi del glucosio nel corpo intero e la sensibilità all’insulina. I topi ChREBPLiverKO mostrano una tolleranza al glucosio peggiorata, mentre sono protetti dalla steatosi epatica. La delezione epatica di ChREBP inoltre ha provocato i cambiamenti di espressione genica nei tessuti adiposi bianchi e marroni, suggerenti la comunicazione inter-tissutale. Il contributo di ChREBP all’equilibrio energetico dell’intero corpo può quindi contare sulla sua regolazione delle specie lipidiche e / o sulla produzione di epatochine che contribuiscono al coordinamento inter tissutale dell’omeostasi energetica (Jois et al., 2017).
Il ChREBP adiposo collega la lipogenesi alla sensibilità all’insulina
La segnalazione alterata dell’insulina nel tessuto adiposo è una caratteristica critica della resistenza all’insulina. Gli studi hanno riferito che l’attivazione di ChREBP nel tessuto adiposo bianco può migliorare l’omeostasi metabolica producendo segnali circolanti protettivi (Yore et al., 2014; Tang et al., 2016). Una classe di lipidi di mammiferi caratterizzati da un legame estere ramificato tra un acido grasso e un acido idrossi-grasso, acido palmitico idrossil stearico (PAHSA), è stata segnalata per esercitare effetti benefici sull’omeostasi del glucosio attraverso la modulazione diretta e incretina-mediata della funzione delle cellule β, l’assorbimento del glucosio e la riduzione dell’infiammazione (Yore et al., 2014). Allo stesso modo, il knockout CHREBP specifico adiposo (ChREBPadiposeKO), che presenta bassi tassi di lipogenesi nel tessuto adiposo, è insulino-resistente con alterata azione insulinica nel fegato, nel muscolo e nel tessuto adiposo bianco sia in condizioni di dieta ricca di grassi che di grassi. I topi ChREBPadiposeKO hanno livelli sierici più bassi di PAHSA, mentre la supplementazione di PAHSA, in particolare l’isomero 9-PAHSA, salva la resistenza all’insulina globale ChREBPadiposeKO e l’infiammazione del tessuto adiposo, confermando che la perdita di grasso-ChREBP è sufficiente a causare insulino-resistenza (Vijayakumar et al., 2017). Un recente studio ha identificato l’obiettivo meccanicistico del complesso rapamicina 2 (mTORC2) come nuovo regolatore di ChREBP (in particolare l’isoforma β) nelle cellule adipose. Ablazione specifica del compagno insensibile alla rapamicina di mTOR (Rictor) negli adipociti maturi compromissione dell’assorbimento di glucosio stimolato dall’insulina nel tessuto adiposo che porta alla down-regolazione di ChREBPß e dell’espressione genica target coinvolta nel controllo della lipogenesi (Tang et al., 2016). In accordo con un’importante diafonia adiposo–epatica mediata da ChREBP, questi effetti sono associati alla resistenza all’insulina epatica e alla gluconeogenesi potenziata. Complessivamente, questi studi supportano un ruolo importante per il ChREBP adiposo nell’innescare segnali insulino-sensibili (Tang et al., 2016).
Nuova interazione tra lipasi sensibile all’ormone e ChREBP nel tessuto adiposo
ChREBP è stato recentemente identificato come partner dell’enzima lipolitico lipasi sensibile all’ormone (HSL) nel tessuto adiposo (Morigny et al., 2019). È stato dimostrato che l’abbattimento di HSL negli adipociti umani e nel tessuto adiposo del topo migliora la sensibilità all’insulina e induce l’allungamento dell’enzima acido grasso a catena molto lunga (Elovl6). Elov16 è un enzima microsomiale che regola l’allungamento degli acidi grassi saturi e monoinsaturi C12-16 in modo ChREBP-dipendente (Morigny et al., 2019). A livello meccanicistico, l’interazione fisica tra HSL e ChREBP ha compromesso la traslocazione nucleare di ChREBPa e la successiva induzione di ChREBPß e geni bersaglio, in particolare Elovl6 (Morigny et al., 2019). Questo studio rivela un nuovo regolamento per ChREBP nel tessuto adiposo. Inibendo l’interazione tra HSL e ChREBP può portare a potenziali strategie terapeutiche per migliorare la sensibilità all’insulina nelle cellule adipose.
Conclusione e direzioni future
ChREBP è ora un sensore di carboidrati ben consolidato. Sebbene la maggior parte degli studi sia stata dedicata alla sua implicazione nel controllo delle vie glicolitiche e lipogeniche, i dati recenti hanno anche svelato nuovi contributi di ChREBP negli epatociti e nelle cellule adipose dove potrebbe essere determinante nella produzione di epatochine e/o lipochine che innescano la diafonia inter-organo. Come discusso, i co-fattori recentemente identificati (modificatori epigenetici) e/o partner (HSL adiposo) in questi tessuti possono anche rappresentare potenziali strategie terapeutiche per la NAFLD e/o per migliorare la sensibilità sistemica all’insulina. Recenti studi hanno anche sostenuto l’importanza del ChREBP nella regolazione del metabolismo del fruttosio e hanno sottolineato la necessità di una migliore comprensione del suo ruolo e della sua regolazione nell’intestino tenue. Infine, l’identificazione di obiettivi specifici e / o sovrapposti di ChREBPa e ChREBPß nei principali tipi di cellule e la determinazione del loro impatto specifico sulla sensibilità all’insulina saranno di particolare importanza nei prossimi anni.
Contributi degli autori
Tutti gli autori elencati hanno dato un contributo sostanziale, diretto e intellettuale al lavoro, e lo hanno approvato per la pubblicazione.
Finanziamento
Il Postic’s lab (U1016-Institut Cochin) è sostenuto da sovvenzioni della Rete ChroME (Marie Curie Skłodowska Action H2020-MSCA-ITN-2015-675610), della Fondazione per la Ricerca Medica (FRM) (DEQ20150331744) e dell’ANR-15-CE14-0026-Epatokind.
Dichiarazione sul conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.