introduktion
Lemmbandet Muskeldystrofier (LGMDs) inkluderar en heterogen grupp av störningar som kännetecknas av progressiv slöseri och svaghet hos de proximala lemmarna (1, 2). LGMDs visar inter-och intrafamilial variabilitet, allt från mycket milda former, till svår, tidig debut, snabbt progressiva fenotyper (2). LGMDs kan klassificeras som autosomal dominant (LGMD1) eller recessiv (LGMD2). Den första gruppen har vanligtvis en vuxen ålder och är inte särskilt vanliga (<10% av alla Lgmd), medan de senare är vanligare (1:15000) (1). Bland recessiva former är LGMD2A (eller kalpainopati) den vanligaste lgmd över hela världen, vilket påverkar cirka 30% av alla lgmd-fall (3). Kliniska signaturer av sjukdom inkluderar tiptoe walking, waddling gång, svårighet att springa och klättra trappor, scapular winging. Gemensamma kontrakturer, Achillessenförkortning, skolios observeras vanligtvis, medan ansikts-och nackmuskler inte påverkas (4). En asymptomatisk Hyperckemi (5-80 gånger CPK normala nivåer) hos unga patienter anses vara ett prekliniskt sjukdomsstadium och kan kvarstå i flera år (1, 5). Åldern för muskelsvaghet uppträder vanligtvis vid 15 år, även om den kan uppstå vid tidigare (<12 år) eller senare (>30 år) åldrar. Sjukdomsprogressionen kan leda till förlust av ambulation, andningsinsufficiens och minskad lungvital kapacitet i de avancerade stadierna (6). Hjärtinvolvering (hjärtrytmstörningar, hjärtledningsstörningar, vänster ventrikulär utstötningsdysfunktion) rapporteras endast ibland (6, 7).
diagnosen kalpainopati bekräftas genom detektering av patogena mutationer i CAPN3 (15q15.1) (4), som kodar för olika alternativt skarvade transkript. Emellertid uttrycks fulllängdsavskriften främst i muskelvävnad (8). Det kodade proteinet (CAPN3) är medlem i icke-lysosomala Ca++-beroende cysteinproteaser familj. I muskler deltar CAPN3 i” sarcomere remodeling”, vilket är viktigt för muskelanpassning och tillväxt som svar på funktionella och metaboliska krav. Hittills har mer än 490 patogena mutationer i hela CAPN3 beskrivits, varav de flesta är singelnukleotidförändringar (4). Mutationer i CAPN3 har associerats med mitokondriella abnormiteter, tillväxtfel, ökad oxidativ stress och sarkomere-desorganisation som helt och hållet bidrar till att muskeln inte kan hålla belastningar och därigenom orsakar myofiberdegenerering och muskelavfall (8). Diagnosen av kalpainopati kan vara utmanande på grund av den genetiska heterogeniteten och icke-specificiteten av kliniskt och instrumentellt mönster. Faktum är att fördelningen av muskelsvaghet/atrofi, hypertrofi/pseudo-hypertrofi och senkontrakt ofta delas med andra lgmd eller neuromuskulära störningar. Muskelbiopsimönster i kalpainopati är i allmänhet också icke-specifikt, allt från milda muskulära avvikelser till allvarliga dystrofa förändringar. Dessutom är immunhistokemiska / biokemiska markörer vanligtvis inte tillförlitliga: calpain-signalen kan vara normal även i närvaro av ett icke-funktionellt protein, och vice versa kan det minskas även i andra muskeldystrofier som skiljer sig från kalpainopati.
det rekommenderas därför att utföra en differentiell diagnos för att ge exakta och pålitliga resultat. För detta ändamål har molekylärgenetiska testmetoder utvecklats för att bekräfta diagnosen kalpainopati, inklusive multigenpanel, omfattande genomisk (exome/genomsekvensering) och enkelgenanalys (direkt sekvensering) (9). Implementeringen av nästa generations sekvensering (NGS) var till hjälp för att generera informativa data som ska tillämpas på diagnostiska, prediktiva eller terapeutiska ändamål (10-12). Ngs-genpaneler är baserade på analysen av en uppsättning gener associerade med en specifik sjukdom eller en grupp relaterade störningar, vilka kännetecknas av genetisk och fenotypisk heterogenitet. Ngs-paneler kan också vara användbara för att detektera blandade eller komplexa fenotyper, som härrör från arv av mer än en genetisk defekt från föräldrar (13). I allmänhet är patienter som presenterar komplexa fenotyper, som är negativa mot de kända mutationerna i specialdesignade diagnostiska genpaneler och kräver en bred differentialdiagnos, berättigade till hela exome-eller genomsekvensering. Hela exome / genomsekvensering är dyrare metoder när det gäller datahantering, tolkning och analytiska kostnader men ökar sannolikheten för att ge en molekylär diagnos till en misstänkt genetisk störning (13). När det gäller LGMDs rekommenderas dedikerade ngs-paneler starkt för att säkerställa höga diagnostiska hastigheter, optimal täckning, känslighet och specificitet för klinisk testning (9). Ngs-paneler representerar därför ett av de bästa systemen för att underlätta differentialdiagnos, identifiera nya orsakande mutationer och klargöra genotyp-fenotypkorrelationer (14, 15).
i detta manuskript rapporteras fallet med en patient som drabbats av LGMD2A, för vilken tillämpningen av NGS-panelen inte bara gjorde det möjligt att bekräfta diagnosen kalpainopati (mutationer i CAPN3) utan också att identifiera en ytterligare ny mutation i LMNA-genen associerad med dilaterad kardiomyopati. Med tanke på dessa resultat utvidgades analysen till probandens familjemedlemmar för att ge en mer omfattande tolkning av analytiska data i förhållande till de patologiska fenotyperna.
Fallpresentation
klinisk karakterisering av Proband och släktingar
sjukdomsuppkomsten i proband hänvisades vid 10 års ålder, när hon rapporterar närvaron av kalvhypertrofi med tåvandring, svårigheter att springa, klättra trappor och stå upp. Symtom visade en långsam men progressiv försämring, med efterföljande involvering av proximal övre extremitet. Vid 13 års ålder upptäcktes för övrigt höga nivåer av CPK (~8000 UI/L), aldrig associerade med myoglobinuri. Successivt genomgick patienten flera neuromuskulära undersökningar vid olika specialiserade centra. Två muskelbiopsier utfördes, som visar klassisk dystrofisk bild (hypertrofiska/atrofiska fibrer, inre kärnor, nekrotiska fibrer, ökning av bindväv) med icke-specifika egenskaper. Som förväntat var immunokemi-och immunoblot-studier ofullständiga, vilket indikerar onormal / reducerad sackarios, sackarios, sarkoglykan, sackarios-dystroglykan och dystrofinsignal endast i nekrotiska fibrer (upprepad immunokemi resulterade normalt, även för laminin); immunoblots för dystrofin och calpain avslöjade normala signaler. Calpain – 3-immunobloten är emellertid känd för att inte vara helt känslig för lgmd2a-diagnos, eftersom 20-30% av fallen visar en normal mängd protein (1, 16-18). På grund av den kliniska presentationen (hyperCKmia, kalvhypertrofi/pseudo-hypertrofi) utesluts först dystrofinopatier. Dessutom avslöjade analys av DMD (Xp21), FKRP (19q13.32) och DYSF (2p13.2) gener inte patogena mutationer. Patienten kom till vår observation vid en ålder av 30, presentera axiell och gördel engagemang både i övre och nedre extremiteterna, med betydande vaggande gång och bevingade scapulae. I nedre extremiteter var svaghet framträdande i quadriceps och glutei muskler som var signifikant ipo-atrofisk, tillsammans med bevis på ”uppenbarligen hypertrofisk” (pseudo-hypertrofi) kalvsmuskler (Figur 1). Kramper, myalgi och krusning observerades inte. Fullständig pneumologisk (spirometri och polysomnografi) och kardiologisk (echografi, 24-timmars EKG Holter-övervakning, hjärt-MR, fullständig kardiologisk fysisk inspektion med målinriktad medicinsk historia och kardiovaskulär reflexanalys) undersökningar avslöjade ingen signifikant abnormitet. Vi utesluter också syra maltasbrist (normala enzymaktivitetsanalyser i lymfocyter/leukocyter). Muskelmagnetisk resonansavbildning (MRI) visade ett framträdande engagemang av scapular bälte, paravertebrala muskler, bakre fackmuskler i låret (med relativ sparsam av sartorius-och gracilismusklerna) och av benet (särskilt gastrocnemius medialis och soleus) (Figur 2). Detta mönster av engagemang har redan rapporterats i litteraturen om MR-bild i kalpainopati (19, 20). Dessutom visade MR-studien att kalvsmusklerna var effektivt atrofiska och kännetecknade av fettinfiltrering, vilket orsakade en muskulös ”utvidgning.”Den kliniska presentationen, startåldern, progressionshastigheten, fördelningen av muskelsvaghet och Mr-resultaten i proband var helt förenliga med en diagnos av LGMD2A som beskrivs i litteraturen (4, 21, 22).
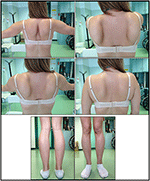
Figur 1. Proband kliniska egenskaper: vingade scapulae och kombination av låratrofi och kalvpseudohypertrofi.
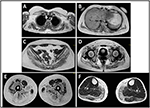
Figur 2. Proband muskel MRI: involvering av övre extremitetsbälte och paravertebrala muskler (A,B), involvering av nedre extremitetsbälte och bakre del av låret (C–E, mestadels glutei) med relativ sparsamhet av sartorius och gracilis, och involvering av benets bakre del (F, mestadels gastrocnemius medialis/soleus).
utvärderingen av familjemedlemmarna i proband avslöjade ingen historia av neuromuskulär sjukdom och inga bevis på konsanguinitet mellan föräldrar. Men probandens mor, vid 55 års ålder, presenterade 2 syncopala episoder för vilka hon diagnostiserades med idiopatisk bradykardi (upp till 40 slag per natt). Omfattande kardiologiska undersökningar på probandens mor diagnostiserade ett symptomatiskt atrioventrikulärt (AV) block efter förekomsten av flera synkopala episoder/lipotymi. Analysen visade närvaron av ett grundläggande första gradens av-block, med flera perioder med svår bradykardi, mestadels nattlig och ofta symptomatisk, associerad med vissa faser av andra gradens av-block, både typ 1 och 2, och vissa faser av nattlig fullständig av-dissociation. Ekokardiografi och ergometriskt test avslöjade inte signifikanta förändringar, med undantag för patentet foramen ovale. Mamman uppvisade inga andra kliniska symptom, predisponerande tillstånd eller riskfaktor. Med tanke på hennes kliniska bild implanterades probandens mor med PMK. Dessutom avslöjade familjehistoria att mormor och farfar till proband också implanterades med PMK i åldern 55 respektive 30 år. Dessutom dog bror till mormor av proband vid 51 för svår dilaterad kardiomyopati. Kardiologiska undersökningar utfördes också på Fadern och moderns farbror till proband som resulterade helt opåverkad.
denna studie godkändes av Santa Lucia-stiftelsens etikutskott och utfördes enligt Helsingforsdeklarationen. Alla deltagare gav undertecknat informerat samtycke för genetisk analys, och i detta avseende gav de också samtycke till publicering av denna fallrapport.
laboratorieundersökningar och diagnostiska tester
genomiskt DNA extraherades från perifert blod (400 kg) med hjälp av MagPurix-DNA-Extraktionssats och MagPurix Automatic Extraction System (Resnova) enligt tillverkarens anvisningar. Prover sekvenserades med användning av Jon PGM-System och Jon Ampliseq Anpassad Panel hög specificitet (Thermo Fisher Scientific). Storleken på panelen var 129.13 Kb, som förväntas screena ~99,72% av den totala panelen med en minsta täckning på 20X. panelen inkluderade 18 gener, som valdes med hjälp av vetenskaplig litteratur, GeneReviews( www.ncbi.nlm.nih.gov/books/NBK1116/) och frekvens av patogena varianter i den allmänna befolkningen. En detaljerad beskrivning av NGS-Panelen har sammanfattats i Tabell 1.
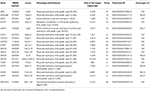
Tabell 1. Anpassad ngs-panel som används för lgmd-diagnos.
Bibliotek konstruktion utfördes av Ion AmpliSeq Bisexual Library Kits 2.0. Ungefärligt 10 ng / Aci l av Start-DNA användes för multiplex-PCR-reaktioner. Successivt utfördes två reningssteg (med AMPure XP, Beckman Coulter) för att avlägsna oönskade föroreningar och en slutlig PCR utfördes. Mallförstärkning och anrikningssteg utfördes av Ion PGM Hi-Q OT2 kit-400, Ion OneTouch 2-systemet och Ion OneTouch ES (Thermo Fisher Scientific). Prover bearbetades av Jon PGM Hi-Q Sekvenseringssats (400 bp, Thermo Fisher Scientific) och kör på Jon 316 Chip v2 (850 flöden krävs) och Jon PGM Sequencer (Thermo Fisher Scientific). Resultaten analyserades med användning av Ion Reporter 4.6 (Thermo Fisher Scientific) och Integrated Genome Viewer (IGV). Tolkningen av genetiska varianter utfördes av Human gen Mutation Database (HGMD), Leiden Open Variation Database (LOVD), ClinVar och ExAC. Den funktionella effekten av de detekterade varianterna utvärderades med bioinformatiska prediktiva verktyg, inklusive Mutationsmakare, Varsome, SIFT, PolyPhen 2, SMART, Human Splicing Finder (HSF). Direkt sekvensering (BigDye Terminator v3.1, BigDyeX Terminator och ABI3130, Applied Biosystems) utfördes för att bekräfta genetiska varianter och för att sekvensera genomiska kodningsregioner med en täckning <20X (LMNA, Chr1:156106052-156106076; DYSF, Chr2:71753352-71753502, Chr2:71776385-71776640; SGCB, Chr4:52904225-52904560; Sgca, chr17:48243242-48243570).
resultat
ngs-analysen av proband avslöjade 3 heterozygota varianter (kompletterande Figur 1). Två varianter lokaliserades i CAPN3, nämligen NM_000070.2 (CAPN3): c.550dela (p.Thr184Argfs) och c.1813g>C (P.Val605Leu) i exonerna 4 respektive 16. Den första är en enda nukleotid deletion och är den vanligaste (75% av fallen) patogena varianten i Europeiska länder (1). Som förväntat klassificerade den bioinformatiska analysen c.550dela (rs80338800) som en funktionsförlustvariant (p.Thr184Argfs), vilket orsakade en ramförskjutning av den öppna läsramen. De prediktiva verktygen (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) beskrev varianten som skadlig. Dessutom avslöjade SMART att den förändrade proteinprodukten saknar calpain 3-domänen och de tre ”EF-hands” – motiven. Den första domänen är involverad i signalvägarna för Calpain medan EF-händerna är väsentliga för CA++-beroende aktivering av proteinet (4).
när det gäller c.1813g>C är det en ny missense-variant (s.Val605Leu) som har förutsagts vara skadlig (genom Mutationsprovning, HSF, Varsome, PolyPhen 2, SIFT) på grund av en potentiell förändring av skarvning. Denna variant är inte kommenterad i litteratur eller bland online-databaser och den har inte hittats i 200 kontrollämnen. Tyvärr gav analysen av SMART tool inte signifikanta resultat, eftersom varianten ligger inom en okarakteriserad domän. Segregeringsanalysen av CAPN3 visade att modern och morbror båda var heterozygota för c.550dela, medan fadern var bärare av c.1813g>C.
dessutom avslöjade ngs-analys på proband närvaron av en ny variant i LMNA, nämligen NM_170707 (LMNA): c.550C>T (p.Gln184*). Den ovan nämnda varianten har förutspåtts vara en nollvariant, har ännu inte beskrivits i litteratur eller bland online-databaser och den har inte hittats i 200 kontrollpersoner. Prediktiva verktyg (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) beskrev en patogen effekt av denna variant på proteinprodukten. SMART tool rapporterade att det förändrade lmna-proteinet saknar filamentdomänen, vilket är viktigt för att bibehålla sin struktur och funktion. Segregeringsanalysen lyfte fram närvaron av c.550C>t-variant endast hos modern, vilket tyder på en möjlig koppling till hennes hjärtsymtomatologi och hennes familjehistoria av sjukdom.
enligt de kriterier som fastställts av American College of Medical Genetics (ACMG) standarder och riktlinjer (23), c.1813g> C (CAPN3) kan beskrivas som en sannolik patogen variant med tanke på att den är belägen i en kritisk domän utan godartad variation (PM1); det är frånvarande i ExAc, GnomAD och 1000 Genome Browser databaser (PM2); med tanke på recessiv arvsmodell har den detekterats i trans med en patogen variant (PM3); flera rader av beräkningsbevis stöder en skadlig effekt på genen eller genprodukten (PP3). När det gäller den kliniska klassificeringen av c.550C>T (LMNA) av ACMG kan den betecknas som en patogen variant, eftersom den är en nollvariant som orsakar funktionsförlust (p.Gln184*) i LMNA (PVS1); den är frånvarande i ExAc, GnomAD och 1000 Genombläddrardatabaser (PM2) ; Den ligger i en kritisk domän utan godartad variation (PM1); flera rader av beräkningsbevis stöder en skadlig effekt på genen eller genprodukten (PP3).
slutsatser
denna fallrapport presenterade en patient som drabbats av LGMD, som befanns vara bärare av mutationer inte bara i CAPN3 (c.550dela och c.1813g>C) utan också i LMNA (c.550C>T). Dessa resultat förklarar probandens neuromuskulära fenotyp på grund av CAPN3-mutationer och belyser en potentiell risk för hjärt-kärlsjukdomar på grund av närvaron av varianten i LMNA och hennes positiva familjehistoria. Med tanke på dessa resultat utsattes familjemedlemmarna för segregeringsanalysen. I synnerhet resulterade moderen att vara bärare av CAPN3_c.550dela respektive LMNA_c.550C>T. Mamman kan betraktas som en frisk bärare för CAPN3_c.550dela patogen mutation associerad med kalpainopati. Närvaron av en ny variant i LMNA kan dock förklara hennes kardiovaskulära patologi (bradykardi och syncopala episoder). Frånvaron av neuromuskulär symptomatologi hos moderen och den speciella kliniska bilden av proband utesluter en möjlig förening av LMNA_c.550C> t med neuromuskulär fenotyp, särskilt när det gäller Emery-Dreifuss muskeldystrofi (EMD). I själva verket påverkar EMD specifikt vastus lateralis och biceps brachii muskler som är relativt skonade i LGM2A (24, 25).
moderns farbror var heterozygot endast för CAPN3_c.550dela och visade som förväntat inga neuromuskulära eller kardiovaskulära problem. Fadern resulterade i att bära romanen CAPN3_c.1813g>C och därmed var han opåverkad. Sammantaget bekräftade segregeringsanalys arvet av probandens tre mutationer från hennes släktingar och lyfte fram en förtrogenhet för kardiomyopati som inte kan försummas (Figur 3).
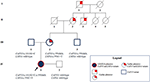
Figur 3. Stamtavla som visar den positiva förtrogenheten för hjärtfenotyp som ärvs av moderns härstamning och överföringen av CAPN3-och LMNA-varianterna i hela familjemedlemmarna.
sammantaget ger dessa data några viktiga överväganden. Först var ngs-panelen i denna patient kritisk för att nå molekylär diagnos av kalpainopati, detektera en känd mutation och en andra ny variant förutsagd som patogen. För det andra tillät ngs-panelen identifieringen av den nya lmna_c.550C>t-varianten i proband. Detta resultat överensstämde med den positiva familjehistorien och med segregeringsdata, förklarar hjärt-kärlsjukdom hos modern och, ännu viktigare, rekommendera mer specifik kardiologisk uppföljning i proband. Det är viktigt att notera att kardiologiska manifestationer inträffade hos modern senare i åldern (55 år), så att vi kan anta att proband fortfarande kan vara i en ”asymptomatisk kardiologisk fas.”Emellertid kan ett variabelt fenotypiskt uttryck av LMNA-mutation i proband jämfört med modern inte uteslutas. Alla dessa data understryker vikten av ett integrerat tillvägagångssätt mellan kliniker och genetiker, för korrekt tolkning av resultat, korrekt genetisk rådgivning och så småningom klinisk hantering och uppföljning. När det gäller genetisk och familjär rådgivning utfördes ngs-analysen också på probandens partner för att uppskatta reproduktionsrisken för paret. Partnern var negativ mot alla de 18 testade generna, vilket innebär att han har 1/650 kvarvarande risk för att vara en hälsosam bärare för lgmd-orsakande mutationer, med tanke på att testet är 84% känsligt. Med hänsyn till probandens genetiska profil och ngs-testets känslighet är den återstående risken för paret att få ett barn som drabbats av kalpainopati 1/1300. Å andra sidan överförs de lmna-patogena varianterna enligt ett autosomalt dominerande mönster, vilket innebär att proband har 50% sannolikhet att ha heterozygota barn. Den kliniska bilden av avkomman kan emellertid inte säkert förutsägas eftersom LMNA_c. 550C>T är en ny variant och dess funktionella inverkan på fenotypen är fortfarande okänd.
Sammanfattningsvis belyser denna fallrapport den kliniska nyttan av NGS-paneler för att ge korrekt lgmd2a-diagnos och beskriva komplexa fenotyper och komorbiditeter som härrör från arv av olika mutationer i flera gener. Tillämpningen av NGS i klinisk praxis bör dock alltid kombineras med en pre – och postgenetisk rådgivning för att ge en tydlig förklaring av resultaten, de möjliga konsekvenserna för patienternas fenotyp, återfallsrisken inom familjen samt för att förklara möjliga oväntade fynd.
datatillgänglighet
inga datauppsättningar genererades eller analyserades för denna studie.
Etikdeklaration
denna studie genomfördes i enlighet med rekommendationerna från ethics committee of Santa Lucia Foundation med skriftligt informerat samtycke från alla ämnen. Alla ämnen gav skriftligt informerat samtycke i enlighet med Helsingforsdeklarationen. Protokollet godkändes av ethics committee of Santa Lucia Foundation.
Författarbidrag
RC, CSt, VC, GC, RG, GPa, SC, CP och JM bidrog till förvärv av data, analys och tolkning av data. CSt, RC, GC, RG, GM och EG har varit inblandade i utarbetandet av manuskriptet. SZ, GPr, CSA och SS har varit involverade i förvärvet av kliniska data. RC, CSt, VC, GC, RG, GPa, SC, CP, JM, SZ, GPr, GM, CSa, SS och EG har gett slutligt godkännande av den version som ska publiceras.
finansiering
detta arbete stöds av 5x 2016 National Health Ministry 2.
intressekonflikt uttalande
författarna förklarar att forskningen genomfördes i avsaknad av kommersiella eller finansiella relationer som kan tolkas som en potentiell intressekonflikt.
tilläggsmaterial
Tilläggsmaterialet för denna artikel finns online på: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. Protein och genetisk diagnos av muskeldystrofi i extremiteterna typ 2a: utbytet och fallgroparna. Muskel Nerv. (2015) 52:163–73. doi: 10.1002 / mus.24682
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
2. Nigro V, Savarese M. genetisk grund för muskeldystrofier i lemmar: 2014-uppdateringen. Acta Myol. (2014) 33:1–12.
PubMed Abstrakt / Google Scholar
3. Han är en av de mest kända och mest kända i världen. Natural history of LGMD2A för att avgränsa resultatmått i kliniska prövningar. Ann Clin Transl Neurol. (2016) 3:248–65. doi: 10.1002 / acn3.287
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
4. Angelini C, Fanin M. Calpainopathy. I: Adam MP, Ardinger HH, Pagon RA redaktörer. GeneReviews. Seattle, WA: University of Washington (2017) 1-30.
PubMed Abstrakt / Google Scholar
5. J. A., J. A., J. A., S. A., Siciliano G., Vilchez J. J., et al. EFNS riktlinjer för diagnostisk metod för pauci – eller asymptomatisk hyperckemi. Eur J Neurol. (2010) 17:767–73. doi: 10.1111 / j. 1468-1331. 2010. 03012.X
CrossRef fulltext / Google Scholar
6. Han är en av de mest kända och mest kända i världen. Kardiopulmonell dysfunktion hos patienter med muskeldystrofi i extremiteten 2A. Muskelnerv. (2017) 55:465–9. doi: 10.1002 / mus.25369
CrossRef Fulltext / Google Scholar
7. Okere A, Reddy SS, Gupta S, Shinnar M. en kardiomyopati hos en patient med muskeldystrofi i extremiteterna typ 2A. Circ hjärtfel. (2013) 6:e12–13. doi: 10.1161 / CIRCHEARTFAILURE.112.971424
CrossRef Fulltext / Google Scholar
8. Kramerova I, Ermolova N, Eskin A, Hevener A, Quehenberger O, Armando AM, et al. Underlåtenhet att reglera transkription av gener som är nödvändiga för muskelanpassning ligger till grund för muskeldystrofi i extremiteterna 2A (kalpainopati). Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093 / hmg / ddw086
PubMed Abstrakt / CrossRef fulltext / Google Scholar
9. Thompson R, Straub v. lem gördel muskeldystrofier-internationella samarbeten för translationell forskning. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038 / nrneurol.2016.35
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
10. Det är en av de viktigaste frågorna för dig. Tillämpning av precisionsmedicin i neurodegenerativa sjukdomar. Främre Neurol. (2018) 9:701. doi: 10.3389/fneur.2018.00701
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
11. Cascella R, Strafella C, Caputo V, Errichiello V, Zampatti S, Milano F, et al. Mot tillämpning av precisionsmedicin vid åldersrelaterad makuladegeneration. Prog Retin Eye Res. (2017) 63:132-46. doi: 10.1016 / j.preteyeres.2017.11.004
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
12. Cascella R, Strafella C, Longo G, Manzo L, Ragazzo M, De Felici C, et al. Bedömning av individuell risk för AMD med genetisk rådgivning, familjehistoria och genetisk testning. Ögon. (2017) 32:446–50. doi: 10.1038 / öga.2017.192
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
13. Adams DR, Eng CM. Nästa generations sekvensering för att diagnostisera misstänkta genetiska störningar. N Engl J Med. (2019) 379:1353–62. doi: 10.1056 / NEJMc1814955
PubMed Abstrakt / CrossRef fulltext / Google Scholar
14. Angelini C. neuromuskulär sjukdom. Diagnos och upptäckt i muskeldystrofi i extremiteterna. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038 / nrneurol.2015.230
CrossRef Fulltext / Google Scholar
15. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, et al. Användning av hel-exome sekvensering för diagnos av lem-gördel muskeldystrofi: resultat och lärdomar. JAMA Neurol. (2015) 72:1424–32. doi: 10.1001 / jamaneurol.2015.2274
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
16. Milic A, Daniele N, Lochm Obbller H, Mora M, Comi GP, Moggio M, et al. En tredjedel av lgmd2a-biopsier har normal calpain 3-proteolytisk aktivitet som bestäms av en in vitro-analys. Neuromusc Disord. (2007) 17:148–56. doi: 10.1016/j.nmd.2006.11.001
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
17. Lanzillo R, Aurino S, Fanin M, Aguennoz M, Vitale F, Fiorillo C, et al. Tidig start calpainopathy med normal icke-funktionell calpain 3 nivå. Dev Med Barn Neurol. (2006) 48:304–6. doi: 10.1017 / S001216220600065X
PubMed Abstrakt / CrossRef fulltext / Google Scholar
18. Talim B, Ognibene A, Mattioli E, Richard i, Anderson LV, Merlini L. normalt kalpainuttryck i genetiskt bekräftad muskeldystrofi av lem-bälte typ 2a. neurologi. (2001) 56:692–3. doi: 10.1212 / wnl.56.5.692-a
CrossRef fulltext / Google Scholar
19. D-manera J, Llauger J, Gallardo E, Illa I. muskel-MR i muskeldystrofier. Acta Myol. (2015) 34:95–108.
PubMed Abstrakt / Google Scholar
20. Det finns många olika typer av produkter. Muskel – Mr-fynd hos patienter med muskeldystrofi i extremiteterna med calpain 3-brist (LGMD2A) och tidiga kontrakturer. Neuromusc Disord. (2005) 15:164–71. doi: 10.1016/j.nmd.2004.10.008
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
21. Magri F, Nigro V, Angelini C, Mongini T, Mora M, Moroni I, et al. Den italienska lem gördel muskeldystrofi registret: relativ frekvens, kliniska egenskaper och differentialdiagnos. Muskel Nerv. (2017) 55:55–68. doi: 10.1002 / mus.25192
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
22. J., J., J., J., N., N., N., M., M., M., M., M., M., M., M., M., M., M., M., M. Calpain 3 genmutationer: genetiska och klinisk-patologiska fynd i muskeldystrofi i extremiteterna. Neuromusc Disord. (2001) 11:547–55. doi: 10.1016 / S0960-8966(01)00197-3
PubMed Abstrakt / CrossRef fulltext / Google Scholar
23. Han är en av de mest kända och mest kända. Standarder och riktlinjer för tolkning av sekvensvarianter: en gemensam konsensusrekommendation från American College of Medical Genetics and genomics och Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038 / gim.2015.30
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
24. Bonne G, Mercuri E, Muchir A, Urtizberea A, B C, Recan D, et al. Kliniskt och molekylärt genetiskt spektrum av autosomalt dominerande Emery-Dreifuss muskeldystrofi på grund av mutationer av lamin A/C-genen. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170::stöd-ANA6>3.0.CO; 2-J
PubMed Abstrakt / CrossRef fulltext / Google Scholar
25. Vi är ett av de mest populära företagen i världen. Hjärtrytmrubbningar, kardiomyopati och muskeldystrofi hos patienter med Emery-Dreifuss muskeldystrofi och lem-gördel muskeldystrofi typ 1B. J koreanska med Sci. (2005) 20:283–90. doi: 10.3346/jkms.2005.20.2.283
CrossRef Fulltext / Google Scholar