- introduktion
- metoder
- filtrering av Ribosomal Profileringsdata
- montering av virtuella genom
- bestämning av Ribo-seq Read-Mapped Region på en korsning (RMRJ) av circRNAs
- utbildning av modellen och klassificering av RMRJs
- förutsägelse av översatta peptider med RMRJs
- resultat och diskussion
- översatta circRNAs hos människor och A. thaliana
- funktionell anrikning av humana och A. thaliana circRNAs med Kodningspotential
- Noggrannhetstest för CircCode
- påverkan av Ribo-seq-Datasekvenseringsdjupet
- jämförelse av CircCode med andra verktyg
- slutsatser
- tillgänglighet och krav
- datatillgänglighet uttalande
- Författarbidrag
- finansiering
- intressekonflikt
- tilläggsmaterial
introduktion
cirkulära RNA (circRNAs) är en speciell typ av icke-kodande RNA-molekyl som har blivit ett hett forskningsämne inom RNA och får stor uppmärksamhet (Chen och Yang, 2015). Jämfört med traditionella linjära RNA (innehållande 5′ och 3′ ändar) har circRNA-molekyler vanligtvis en sluten cirkulär struktur; vilket gör dem mer stabila och mindre benägna att nedbrytas (Vicens och Westhof, 2014). Även om förekomsten av circRNAs har varit känd under en tid, ansågs dessa molekyler vara en biprodukt av RNA-Splitsning. Men med utvecklingen av sekvensering med hög genomströmning och bioinformatikteknik har circRNAs blivit allmänt erkända hos djur och växter (Chen och Yang, 2015). Nya studier har också visat att ett stort antal circRNAs kan översättas till små peptider i celler (Pamudurti et al., 2017) och har nyckelroller trots deras ibland låga uttrycksnivå (Hsu och Benfey, 2018; Yang et al., 2018). Även om ett ökande antal circRNAs identifieras, återstår deras funktioner i växter och djur i allmänhet att studeras. Förutom deras funktioner som miRNA-lockbete har circRNAs viktig translationspotential, men inga verktyg finns tillgängliga för att specifikt förutsäga translationsförmågan hos dessa molekyler (Jakobi och Dieterich, 2019).
flera verktyg finns för förutsägelse och identifiering av circRNAs, såsom CIRI (Gao et al., 2015), CIRCexplorer (Dong et al., 2019), CircPro (Meng et al., 2017), och circtools (Jakobi et al., 2018). Bland dem kan CircPro avslöja översatta circRNAs genom att beräkna en översättningspotentialpoäng för circRNAs baserat på CPC (Kong et al., 2007), som är ett verktyg för att identifiera open reading frame (ORF) i en given sekvens. Men eftersom vissa circRNAs inte använder startkodonet under översättning (Ingolia et al., 2011; Slavoff et al., 2013; Kearse och Wilusz, 2017; Spealman et al., 2018), anställa CPC kan filtrera bort några verkligt översatta circRNAs. I denna studie använde vi BASiNET (ito et al., 2018), som är en RNA-klassificerare baserad på maskininlärningsmetoderna (random forest och J48-modellen). Den omvandlar initialt de givna kodande RNA (positiva data) och icke-kodande RNA (negativa data) och representerar dem som komplexa nätverk; den extraherar sedan de topologiska måtten på dessa nätverk och konstruerar en funktionsvektor för att träna modellen som används för att klassificera kodningskapaciteten för circrna. Med denna metod undviks felaktig filtrering av översatta circRNAs som inte initieras av AUG. Dessutom Ribo-seq-teknik, som är baserad på sekvensering med hög genomströmning för att övervaka RPFs (ribosomala skyddade fragment) av transkript (Guttman et al., 2013; Brar och Weissman, 2015), kan användas för att bestämma platserna för circRNAs som översätts (Michel och Baranov, 2013). För att identifiera circrnas kodningsförmåga utvecklade vi verktyget CircCode, som involverar en Python 3–baserad ram, och tillämpade CircCode för att undersöka översättningspotentialen för circRNAs från människor och Arabidopsis thaliana. Vårt arbete ger en rik resurs för vidare studier av circRNAs funktioner med kodningskapacitet.
metoder
CircCode skrevs i Python 3 programmeringsspråk; den använder Trimmomatic (Bolger et al., 2014), bowtie (Langmead och Salzberg, 2012) och STAR (Dobin et al., 2013) för att filtrera rå Ribo-seq läser och kartlägger dessa filtrerade läser till genomet. CircCode identifierar sedan Ribo-seq lästa mappade regioner i circRNAs som innehåller korsningar. Därefter sorteras de kandidatmappade sekvenserna i circrna baserat på klassificerare (J48-modell) i kodande RNA och icke-kodande RNA av BASiNET. Slutligen identifieras korta peptider som produceras genom översättning som potentiella kodande regioner av circRNAs. Hela processen med Circode består av fem steg (Figur 1).
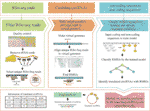
Figur 1 arbetsflödet för CircCode. Det översta lagret representerar indatafilen som krävs för varje steg i CircCode. Mellanskiktet är uppdelat i tre delar, och varje del representerar ett annat driftsstadium. Från vänster till höger representerar den första delen filtreringen av Ribo-seq-data; kvalitetskontrollen utförs av Trimmomatic, och rRNA-avläsningarna avlägsnas av bowtie. Den andra delen representerar stegen som används för att producera det virtuella genomet och anpassa de filtrerade läsningarna till det virtuella genomet med stjärna. Den sista delen representerar identifieringen av översatta circRNAs genom maskininlärning. Bottenskiktet representerar det sista steget som används för att förutsäga peptiderna översatta från circrna och de slutliga resultaten, inklusive information om översatta circrna och deras översättningsprodukter.
filtrering av Ribosomal Profileringsdata
först avlägsnas fragment och Adaptrar av låg kvalitet i Ribo-Seq-avläsningarna av Trimmomatic med standardparametrarna för att få rena Ribo-seq-läsningar. För det andra mappas dessa rena Ribo-seq-läsningar till ett rRNA-bibliotek för att ta bort läsningar som härrör från rRNA med bowtie. Eftersom läslängderna för Ribo-seq är relativt korta (vanligtvis mindre än 50 bp) är det möjligt för en läsning att matcha flera regioner. I det här fallet är det svårt att bestämma vilken region en viss läsning motsvarar. För att undvika detta kartläggs de rena Ribo-seq-läsningarna till genomet av en art av intresse, och de läsningar som inte är perfekt anpassade till genomet betraktas som den slutliga unika Ribo-seq-läsningen.
montering av virtuella genom
CircRNAs visas vanligtvis som ringformade molekyler i eukaryoter, och de kan identifieras baserat på deras back-splicing korsningar. Sekvenserna av circRNAs i fasta-filen är emellertid ofta i linjär form. I teorin indikerar resultatet att korsningen är mellan 5′-terminalnukleotiden och 3′ – terminalnukleotiden, även om korsningen och sekvensen nära korsningen inte kan ses direkt, vilket således anpassar Ribo-seq läser till circRNA-sekvenser, inklusive korsningar, på ett enkelt sätt.
CircCode förbinder sekvensen för varje circRNA i tandem så att korsningen för varje är i mitten av den nybyggda sekvensen. Vi separerade också varje serieenhet med 100 N nukleotider för att undvika förvirring vid sekvensinriktningssteget (längden på varje RPF är mindre än 50 bp). Slutligen erhöll vi ett virtuellt genom som endast består av kandidatcirkrar i tandem åtskilda av 100 Ns. Eftersom CircCode endast fokuserar på anpassning mellan Ribo-seq läser och circRNA sekvenser, kan vi undersöka kodningspotentialen för circRNAs genom att kartlägga Ribo-seq läser till detta virtuella genom, vilket kan spara en stor mängd beräkningstid (det virtuella genomet är mycket mindre än hela genomet) och öka noggrannheten (genom att undvika störningar mellan uppströms och nedströms sekvensjämförelser av circRNAs).
bestämning av Ribo-seq Read-Mapped Region på en korsning (RMRJ) av circRNAs
den slutliga unika Ribo-seq läser mappas till ett tidigare skapat virtuellt genom med hjälp av stjärna. Eftersom varje tandem circRNA-enhet separerades av 100 N-baser innan det virtuella genomet producerades, sattes den största intron-längden till att inte överstiga 10–baser med parametern ” – alignIntronMax 10.”Denna parameter eliminerar all interaktion mellan olika circRNAs i sekvensinriktningen. I det andra steget av virtuell genomproduktion lagrar CircCode positionell korsningsinformation för varje circRNA i det virtuella genomet. Om Ribo-seq read-mapped region i det virtuella genomet inkluderar korsningen av circRNA, och antalet mappade Ribo-seq läser på korsningen (NMJ) är större än 3, Ribo-seq reads-mapped region på korsningen av circRNAs kan betraktas som en RMRJ, vilket avslöjar ett grovt översatt segment av circRNAs nära korsningsplatsen.
utbildning av modellen och klassificering av RMRJs
även om RMRJs kan utgöra ett kraftfullt bevis på översättning, finns det fortfarande vissa brister i denna metod. Eftersom längden på avläsningarna av den ribosomala kartan är kort kan en läsning jämföras med fel position. Därför är det inte övertygande att bara betrakta den region som omfattas av Ribo-seq som den översatta regionen. För detta ändamål används maskininlärningsmetoden för att identifiera rmrjs kodningsförmåga. För det första extraherar Circode kodande RNA (positiva data) och icke-kodande RNA (negativa data) från en art av intresse och använder dem för modellutbildning med hjälp av skillnaden i funktionsvektorer mellan kodande och icke-kodande RNA. CircCode använder sedan den utbildade modellen för att klassificera RMRJs som erhållits i föregående steg av BASiNET. Om RMRJ för ett circRNA känns igen som kodande RNA, kan detta circRNA identifieras som ett översatt circRNA.
förutsägelse av översatta peptider med RMRJs
eftersom uttrycket av circRNAs i organismer är lågt visar Ribo-seq-data inte den exakta 3-nt-periodiciteten tydligt när det gäller färre RPFs. Därför är det svårt att bestämma den exakta översättningsstartplatsen för en översatt circRNA. På grund av närvaron av ett stoppkodon i vissa RMRJs och eftersom startkodonet är svårt att bestämma är metoden att hitta en ORF baserad på ett startkodon och ett stoppkodon inte genomförbart.
för att bestämma de verkliga översättningsregionerna för dessa circRNAs och generera den slutliga översättningsprodukten, FragGeneScan (Rho et al., 2010), som kan förutsäga proteinkodande regioner i fragmenterade gener och gener med frameshifts, används för att bestämma de översatta peptiderna som produceras av circRNAs.
för att undvika den besvärliga körprocessen kan alla modeller anropas av ett skalskript; användaren kan helt enkelt fylla i den givna konfigurationsfilen och mata in den i skript, och hela processen för att förutsäga de översatta circrna kommer sedan att köras. Dessutom kan CircCode köras separat, steg för steg, så att användaren kan justera parametrarna mitt i proceduren och se resultaten från varje steg efter önskemål.
resultat och diskussion
efter testning på flera datorer visade sig CircCode köras med de nödvändiga beroenden installerade. För att testa prestanda för CircCode använde vi data för människor och A. thaliana för att förutsäga circRNAs med översättningspotential. Resultaten jämfördes med circRNAs som har verifierats experimentellt som bekräftelse. Därefter testade vi false discovery rate (FDR) värdet av CircCode ytterligare. Vi använde GenRGenS (Ponty et al., 2006) för att generera en datamängd för testning baserad på kända översatta circrna och bekräftade att FDR-värdet låg inom ett acceptabelt intervall och på en låg nivå. Slutligen utvärderade vi effekten av olika sekvenseringsdjup av Ribo-seq-data på Circode-förutsägelser och jämförde Circode med annan programvara.
översatta circRNAs hos människor och A. thaliana
för att tillämpa CircCode-verktyget på verkliga data laddade vi först ner filerna inklusive human reference genome GRCh38, genome annotation och human rRNA, från Ensembl. För A. thaliana hämtades referensgenomerna (TAIR10), genomanteckningsfiler och motsvarande rRNA-sekvenser från Ensembl-växter. Ribo – seq-data för människor och A. thaliana laddades ner från RPFdb (anslutningsnummer: GSE96643, GSE81295, GSE88794) (Hsu et al., 2016; Willems et al., 2017), och alla kandidat circRNAs från human och A. thaliana hämtades från CIRCPedia v2 (Dong et al., 2018) respektive PlantcircBase (Chu et al., 2017). I slutändan identifierade vi 3 610 översatta circRNAs från mänskliga och 1 569 översatta circRNAs från A. thaliana med hjälp av CircCode (kompletterande Data 1).
funktionell anrikning av humana och A. thaliana circRNAs med Kodningspotential
med hjälp av Circode-resultaten för human och A. thaliana, onlineverktyget KOBAS 3.0 (Wu et al., 2006) användes för att kommentera dessa översatta circRNAs baserat på deras modergener. Dessutom utförde vi Go (Gen ontologi) funktionell analys och KEGG (Kyoto Encyclopedia of gener och genomer) anrikningsanalys för dessa översatta circRNAs med användning av r-paketet clusterProfiler (Yu et al., 2012).
KEGG-resultaten visade att de humana circrna berikades i proteinbehandling i endoplasmatisk retikulumväg, kolmetabolismväg och RNA-transportväg. GO-analys indikerade deltagandet av humana översatta circRNAs i regleringen av molekylbindning, ATPas-aktivitet och andra RNA-splitsningsrelaterade biologiska processer. Dessutom är de översatta circRNAs av A. thaliana berikade i vägar relaterade till stressmotstånd, vilket tyder på att de spelar viktiga roller i denna process (kompletterande Data 2).
Noggrannhetstest för CircCode
för att undersöka noggrannheten hos CircCode användes testsekvenser genererade av GenRGenS, som använder den dolda Markov-modellen för att producera sekvenser som har samma sekvensegenskaper (såsom frekvenserna för olika nukleotider, olika kodoner och olika nukleotider i början av sekvensen).
för denna studie använde vi tidigare publicerade mänskliga översatta circRNAs (Yang et al., 2017) som ingång för GenRGenS och genererade 10 000 sekvenser för att testa CircCode. Vi upprepade testet 10 gånger, och i genomsnitt förutspåddes 27 översatta circRNAs varje gång. FDR-värdet beräknades vara 0,0027, vilket är mycket mindre än 0,05, vilket indikerar att de förutsagda resultaten är trovärdiga.
dessutom jämförde vi de översatta circRNAs från människor som identifierats av CircCode med verifierade polysomassocierade circRNA-data (Yang et al., 2017). Bland dem identifierades 60% av circrna med CircCode (kompletterande Data 3).
påverkan av Ribo-seq-Datasekvenseringsdjupet
för att undersöka effekten av sekvenseringsdjupet för Ribo-seq-data på CircCode-identifieringsresultaten testade vi först effekten av sekvenseringsdjupet på antalet översatta circRNAs (figur 2a). När sekvenseringsdjupet var lågt var det förutsagda antalet översatta circRNAs lågt och antalet översatta circRNAs ökade med ökande sekvenseringsdjup. Antalet översatta circRNAs blev stabilt när sekvenseringsdjupet nådde inte mindre än 10 linjär transkripttäckning.

Figur 2 (A) Effekt av Ribo-seq-datasekvenseringsdjup på det förutsagda antalet översatta circRNAs. (B) effekten av junction read number (JRN) på Circode-känslighet vid olika sekvenseringsdjup.
för det andra, påverkan av NMJ på känslighet vid olika sekvenseringsdjup bedömdes också (Figur 2B). Resultaten visade att NMJ hade mindre inverkan på känsligheten när sekvenseringsdjupet ökade. CircCode hade också högre känslighet vid användning av Ribo-seq-data med högre sekvenseringsdjup.
jämförelse av CircCode med andra verktyg
för att jämföra CircCode med andra verktyg, såsom CircPro, samma uppsättning Ribo-seq-data (SRR3495999) från A. thaliana användes för att identifiera översatta circRNAs med sex processorer, med 16 gigabyte RAM. CircPro identifierade 44 översatta circRNAs i 13 min, medan CircCode identifierade 76 översatta circRNAs i 20 min. Således är CircCode känsligare än CircPro på samma datorhårdvarunivå, men det tar mer tid. CircPro är kortfattad och mindre tidskrävande än CircCode, men CircCode kan identifiera fler circRNAs med kodningsförmåga än CircPro.
slutsatser
CircRNAs spelar en viktig roll i biologi, och det är avgörande att exakt identifiera circRNAs med kodningsförmåga för efterföljande forskning. Baserat på Python 3 utvecklade vi CircCode, ett lättanvänt kommandoradsverktyg som har hög känslighet för att identifiera översatta circRNAs från Ribo-Seq läser med hög noggrannhet. CircCode uppvisar bra prestanda i både växter och djur. Framtida arbete kommer att lägga nedströms teckenanalys till CircCode genom att visualisera varje steg i processen och optimera noggrannheten i förutsägelsen.
tillgänglighet och krav
CircCode finns på https://github.com/PSSUN/CircCode; operativsystem(er): Linux, programmeringsspråk: Python 3 och R; andra krav: bedtools (version 2.20.0 eller senare), bowtie, STAR, Python 3 paket (Biopython, Pandas, rpy2), R-paket (BASiNET, Biostrings). Installationspaketen för all nödvändig programvara finns på Circcodes hemsida. Användare behöver inte ladda ner dem individuellt. Circcodes hemsida innehåller också detaljerade användarmanualer för referens. Verktyget är fritt tillgängligt. Det finns inga begränsningar för användning av nonacademics.
datatillgänglighet uttalande
alla relevanta data finns i manuskriptet och dess stödjande informationsfiler.
Författarbidrag
konceptualisering: PS, GL. Data Curation: PS, GL. Formell analys: PS, GL. Skriva-Original utkast: PS, GL. Skriva-granska och redigera: PS, GL.
finansiering
detta arbete stöddes av bidrag från National Natural Science Foundation of China (grant nos. 31770333, 31370329 och 11631012), programmet för New Century Excellent Talents in University (NCET-12-0896) och Grundforskningsfonderna för de centrala universiteten (no. GK201403004). Finansieringsorganen hade ingen roll i studien, dess utformning, datainsamling och analys, beslutet att publicera eller utarbetandet av manuskriptet. Finansiärerna hade ingen roll i studiedesign, datainsamling och analys, beslut att publicera eller förberedelse av manuskriptet.
intressekonflikt
författarna förklarar att forskningen genomfördes i avsaknad av kommersiella eller finansiella relationer som kan tolkas som en potentiell intressekonflikt.
tilläggsmaterial
Tilläggsmaterialet för denna artikel finns online på: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
kompletterande Data 1 / sekvensen för den förutsagda översatta circRNA och korta peptiden.
kompletterande Data 2 / Go anrikning och KEGG anrikning resultat för människor och Arabidopsis thaliana.
kompletterande Data 3 | jämförelse av förutsagda översatta circRNAs med validerade översatta circRNAs.
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: en flexibel trimmer för illumina sekvensdata. Bioinformatik 30, 2114-2120. doi: 10.1093 / bioinformatik / btu170
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Brar, G. A., Weissman, J. S. (2015). Ribosomprofilering avslöjar vad, när, var och hur proteinsyntesen. Nat. Pastor Mol. Cell Biol. 16, 651–664. doi: 10.1038 / nrm4069
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Chen, L.-L., Yang ,L. (2015). Reglering av circRNA biogenes. RNA Biol. 12, 381–388. doi: 10.1080/15476286.2015.1020271
PubMed Abstrakt / CrossRef fulltext / Google Scholar
chu, Q., Zhang, X., Zhu, X., Liu, C., Mao, L., Ye, C., et al. (2017). PlantcircBase: en databas för växtcirkulära rna. Mol. Växt 10, 1126-1128. doi: 10.1016/j.molp.2017.03.003
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). Stjärna: ultrasnabb universal RNA-seq aligner. Bioinformatik 29, 15-21. doi: 10.1093 / bioinformatik / bts635
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Dong, R., Ma, X.-K., Chen, L.-L., Yang, L. (2019). ”Genombrett annotering av circRNAs och deras alternativa backsplicing/splicing med CIRCexplorer Pipeline”, i Epitranscriptomics. EDS. Wajapeyee, N., Gupta, R. (New York, NY: Springer New York), 137-149. doi: 10.1007/978-1-4939-8808-2_10
CrossRef fulltext / Google Scholar
Dong, R., Ma, X.-K., Li, G.-W., Yang, L. (2018). CIRCpedia v2: en uppdaterad databas för omfattande cirkulär RNA-annotering och uttrycksjämförelse. Genomics Proteomics Bioinf. 16, 226–233. doi: 10.1016/j.gpb.2018.08.001
CrossRef Fulltext / Google Scholar
Gao, Y., Wang, J., Zhao, F. (2015). CIRI: en effektiv och opartisk algoritm för de novo cirkulär RNA-identifiering. Genome Biol. 16, 4. doi: 10.1186 / s13059-014-0571-3
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Guttman, M., Russell, P., Ingolia, N. T., Weissman, J. S., Lander, E. S. (2013). Ribosomprofilering ger bevis för att stora icke-kodande RNA inte kodar för proteiner. Cell 154, 240-251. doi: 10.1016/j.cell.2013.06.009
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
Hsu, P. Y., Benfey, P. N. (2018). Liten men mäktig: funktionella peptider kodade av små ORF i växter. Proteomik 18, 1700038. doi: 10.1002/pmic.201700038
CrossRef fulltext / Google Scholar
Hsu, P. Y., Calviello, L., Wu, H.-Y. L., Li, F.-W., Rothfels, C. J., Ohler, U., et al. (2016). Superupplösning ribosomprofilering avslöjar oannoterade översättningshändelser i Arabidopsis. Proc. Natl. Acad. Sci. 113, E7126-E7135. doi: 10.1073/pnas.1614788113
CrossRef Fulltext / Google Scholar
Ingolia, N. T., Lareau, L. F., Weissman, J. S. (2011). Ribosomprofilering av embryonala stamceller från mus avslöjar komplexiteten och dynamiken hos däggdjursproteomer. Cell 147, 789-802. doi: 10.1016/j.cell.2011.10.002
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Ito, E. A., Katahira, I., Vicente, F. F., da, R., Pereira, L. F. P., Lopes, F. M. (2018). BASiNET-BiologicAl Sequences NETwork: en fallstudie om kodning och icke-kodande RNA-identifiering. Nukleinsyror Res. 46, e96-e96. doi: 10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093/bioinformatik / bty948
CrossRef fulltext / Google Scholar
Kearse, M. G., Wilusz, J. E. (2017). Icke-AUG översättning: en ny start för proteinsyntes i eukaryoter. Gener Dev. 31, 1717–1731. doi: 10.1101/gad.305250.117
PubMed Abstrakt / CrossRef fulltext / Google Scholar
L., Zhang, Y., Ye, Z.-Q., Liu, X.-Q., Zhao, S.-Q., Wei, L., et al. (2007). CPC: bedöma proteinkodningspotentialen för transkript med hjälp av sekvensfunktioner och stödvektormaskin. Nukleinsyror Res. 35, W345-W349. doi: 10.1093/nar / gkm391
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Langmead, B., Salzberg, S. L. (2012). Snabb gapped-läs anpassning med fluga 2. Nat. Metoder 9, 357-359. doi: 10.1038 / nmeth.1923
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
Meng, X., Chen, Q., Zhang, P., Chen, M. (2017). CircPro: ett integrerat verktyg för identifiering av circRNAs med proteinkodningspotential. Bioinformatik 33, 3314-3316. doi: 10.1093 / bioinformatik / btx446
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Michel, A. M., Baranov, P. V. (2013). Ribosomprofilering: en Hi-Def-monitor för proteinsyntes i genomskala: ribosomprofilering. Wiley Interscip. Rev. RNA 4, 473-490. doi: 10.1002 / wrna.1172
PubMed Abstrakt / CrossRef fulltext / Google Scholar
han är en av de mest kända och mest kända i världen. (2017). Översättning av CircRNAs. Mol. Cell 66, 9-21.e7. doi: 10.1016 / j.molcel.2017.02.021
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
Ponty, Y., Termier, M., Denise ,A. (2006). GenRGenS: programvara för att generera slumpmässiga genomiska sekvenser och strukturer. Bioinformatik 22, 1534-1535. doi: 10.1093 / bioinformatik / btl113
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Rho, M., Tang, H., Ye, Y. (2010). FragGeneScan: förutsäga gener i korta och felaktiga läsningar. Nukleinsyror Res. 38, e191-e191. doi: 10.1093/nar / gkq747
PubMed Abstrakt / CrossRef fulltext / Google Scholar
han är en av de mest kända och mest kända i världen. (2013). Peptidomisk upptäckt av korta öppna läsramkodade peptider i humana celler. Nat. Chem. Biol. 9, 59–64. doi: 10.1038 / nchembio.1120
PubMed Abstrakt / CrossRef fulltext / Google Scholar
han är en av de mest kända och mest kända i världen. (2018). Konserverade icke-AUG uORFs avslöjade av en ny regressionsanalys av ribosomprofileringsdata. Genom Res. 28, 214-222. doi: 10.1101 / gr.221507.117
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
Vicens, Q., Westhof, E. (2014). Biogenes av cirkulära RNA. Cell 159, 13-14. doi: 10.1016/j.cell.2014.09.005
PubMed Abstrakt / CrossRef fulltext / Google Scholar
han är en av de mest kända och mest kända i världen. (2017). N-terminal proteomics hjälpte profilering av det outforskade översättningsinitieringslandskapet i Arabidopsis thaliana. Mol. Cell. Proteomik 16, 1064-1080. doi: 10.1074 / mcp.M116. 066662
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
Wu, J., Mao, X., Cai, T., Luo, J., Wei, L. (2006). KOBAS server: en webbaserad plattform för automatiserad annotering och pathway identification. Nukleinsyror Res. 34, W720-W724. doi: 10.1093 / nar / gkl167
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Yang, L., Fu, J., Zhou, Y. (2018). Cirkulära RNA och deras framväxande roller i immunreglering. Front. Immunol. 9, 2977. doi: 10.3389 / fimmu.2018.02977
PubMed Abstrakt / CrossRef fulltext / Google Scholar
Yang, Y., Fan, X., Mao, M., sång, X., Wu, P., Zhang, Y., et al. (2017). Omfattande översättning av cirkulära RNA drivna av N6-metyladenosin. Cell Res. 27, 626-641. doi: 10.1038 / cr.2017.31
PubMed Abstrakt / CrossRef Fulltext / Google Scholar
Yu, G., Wang, L.-G., Han, Y., Han, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar