Introducere
distrofiile musculare ale brâului membrelor (LGMDs) includ un grup heterogen de tulburări caracterizate prin irosirea progresivă și slăbiciunea mușchilor proximali ai brâului membrelor (1, 2). Lgmd-urile prezintă variabilitate inter și intrafamilială, variind de la forme foarte ușoare, până la fenotipuri severe, cu debut precoce, rapid progresive (2). Lgmd-urile pot fi clasificate ca autosomal dominant (LGMD1) sau recesiv (LGMD2). Primul grup are de obicei o vârstă adultă de debut și nu sunt foarte frecvente (<10% din toate LGMDs), în timp ce acestea din urmă sunt mai frecvente (1:15000) (1). Printre formele recesive, LGMD2A (sau calpainopatia) este cel mai frecvent LGMD la nivel mondial, afectând aproximativ 30% din toate cazurile LGMDs (3). Semnăturile clinice ale bolii includ mersul pe vârfuri, mersul waddling, dificultatea de a alerga și de a urca scările, aripile scapulare. Contracturile articulare, scurtarea tendonului lui Ahile, scolioza sunt frecvent observate, în timp ce mușchii feței și gâtului nu sunt afectați (4). O hipercemie asimptomatică (de 5-80 ori niveluri normale CPK) la pacienții tineri este considerată o etapă preclinică a bolii și poate persista câțiva ani (1, 5). Vârsta de debut a slăbiciunii musculare apare de obicei la 15 ani, deși poate apărea la vârste mai devreme (<12 ani) sau mai târziu (>30 de ani). Progresia bolii poate duce la pierderea ambulației, insuficiență respiratorie și capacitate vitală pulmonară redusă în stadiile avansate (6). Afectarea cardiacă (tulburări de ritm cardiac, tulburări de conducere cardiacă, disfuncție de ejecție a ventriculului stâng) este raportată doar ocazional (6, 7).
diagnosticul calpainopatiei este confirmat de detectarea mutațiilor patogene în CAPN3 (15q15.1) (4), codificând diferite transcrieri alternativ îmbinate. Cu toate acestea, transcrierea pe toată lungimea este exprimată în principal în țesutul muscular (8). Proteina codificată (CAPN3) este un membru al non-lizozomal ca++-dependent cisteină proteaze familie. În mușchi, CAPN3 participă la” remodelarea sarcomerului”, care este esențială pentru adaptarea și creșterea musculară ca răspuns la cerințele funcționale și metabolice. Până în prezent, au fost descrise mai mult de 490 de mutații patogene în întreaga CAPN3, dintre care majoritatea sunt modificări cu un singur nucleotid (4). Mutațiile din CAPN3 au fost asociate cu anomalii mitocondriale, insuficiență de creștere, stres oxidativ crescut și dezorganizare sarcomere care contribuie în totalitate la a face mușchiul incapabil să dețină SARCINI, provocând astfel degenerarea miofibrei și irosirea musculară (8). Diagnosticul calpainopatiei poate fi dificil din cauza eterogenității genetice și a nespecificității modelului clinic și instrumental. Într-adevăr, distribuția slăbiciunii musculare/atrofiei, hipertrofiei/pseudo-hipertrofiei și contracturilor tendonului sunt foarte des împărtășite cu alte LGMDs sau tulburări neuromusculare. Modelul biopsiei musculare în calpainopatie este, în general, nespecific, variind de la anomalii musculare ușoare până la modificări distrofice severe. Mai mult, markerii imunohistochimici/biochimici nu sunt de obicei fiabili: semnalul calpain poate fi normal chiar și în prezența unei proteine nefuncționale și invers poate fi redus chiar și în alte distrofii musculare diferite de calpainopatie.
prin urmare, se recomandă efectuarea unui diagnostic diferențial pentru a oferi rezultate precise și fiabile. În acest scop, au fost dezvoltate abordări de testare genetică moleculară pentru a confirma diagnosticul de calpainopatie, inclusiv panoul multigen, genomic extensiv (secvențierea exomului/genomului) și analiza cu o singură genă (secvențierea directă) (9). Implementarea secvențierii de generație următoare (NGS) a fost utilă pentru a genera date informative care să fie aplicate în scopuri diagnostice, predictive sau terapeutice (10-12). Panourile genetice NGS se bazează pe analiza unui set de gene asociate cu o boală specifică sau cu un grup de tulburări conexe, caracterizate prin eterogenitate genetică și fenotipică. Panourile NGS pot fi, de asemenea, utile pentru detectarea fenotipurilor amestecate sau complexe, care rezultă din moștenirea mai multor defecte genetice de la părinți (13). În general, pacienții care prezintă fenotipuri complexe, care sunt negativi la mutațiile cunoscute în panourile genetice de diagnostic personalizate și care necesită un diagnostic diferențial larg, sunt eligibili pentru secvențierea întregului exom sau a genomului. Secvențierea întregului exom / genom sunt abordări mai costisitoare în ceea ce privește gestionarea datelor, interpretarea și costurile analitice, dar cresc probabilitatea de a oferi un diagnostic molecular unei tulburări genetice suspectate (13). În ceea ce privește LGMDs, panourile NGS dedicate sunt foarte recomandate, pentru a asigura rate ridicate de diagnostic, acoperire optimă, sensibilitate și specificitate a testelor clinice (9). Panourile NGS reprezintă, prin urmare, unul dintre cele mai bune sisteme pentru a facilita diagnosticul diferențial, a identifica noi mutații cauzale și a clarifica corelațiile genotip-fenotip (14, 15).
în acest manuscris, este raportat cazul unui pacient afectat de LGMD2A, pentru care aplicarea panoului NGS a permis nu numai confirmarea diagnosticului de calpainopatie (mutații în CAPN3), ci și identificarea unei mutații suplimentare, noi, în gena LMNA asociată cu cardiomiopatie dilatativă. Având în vedere aceste rezultate, analiza a fost extinsă la membrii familiei probandului pentru a oferi o interpretare mai cuprinzătoare a datelor analitice în raport cu fenotipurile patologice.
prezentarea cazului
caracterizarea clinică a probandului și a rudelor
debutul bolii în proband a fost menționat la vârsta de 10 ani, când a raportat prezența hipertrofiei gambei cu mersul pe vârfuri, dificultăți în alergare, urcarea scărilor și ridicarea în picioare. Simptomele au arătat o agravare lentă, dar progresivă, cu implicarea ulterioară a membrului superior proximal. La vârsta de 13 ani, niveluri ridicate de CPK (~8.000 UI/L) au fost descoperite întâmplător, niciodată asociate cu mioglobinuria. Succesiv, pacientul a fost supus mai multor investigații neuromusculare la diferite centre specializate. Au fost efectuate două biopsii musculare, prezentând o imagine distrofică clasică (fibre hipertrofice/atrofice, nuclee interne, fibre necrotice, creșterea țesutului conjunctiv) cu caracteristici nespecifice. Așa cum era de așteptat, studiile de imunochimie și imunoblot au fost neconcludente, indicând valori anormale / reduse ale sarcoglicanului de la numărul de la numărul de la numărul de la numărul de la numărul de la numărul de la numărul de la numărul de la numărul de la numărul de la numărul de la numărul de la numărul de la numărul de; imunobloturile pentru distrofină și calpain au evidențiat semnale normale. Cu toate acestea, se știe că imunoblotul calpain-3 nu este complet sensibil pentru diagnosticul LGMD2A, deoarece 20-30% din cazuri prezintă o cantitate normală de proteine (1, 16-18). Datorită prezentării clinice (hiperckmie, hipertrofie de vițel/pseudo-hipertrofie), distrofinopatiile au fost în primul rând excluse. În plus, analiza genelor DMD (Xp21), FKRP (19q13.32) și disf (2p13.2) nu a evidențiat mutații patogene. Pacientul a venit la observația noastră la vârsta de 30 de ani, prezentând implicare axială și brâu atât la nivelul membrelor superioare, cât și la nivelul membrelor inferioare, cu mers semnificativ de waddling și scapule înaripate. În membrele inferioare, slăbiciunea a fost proeminentă în cvadriceps și mușchii glutei care au fost semnificativ ipo-atrofici, împreună cu dovezile mușchilor gambei „aparent hipertrofici” (pseudo-hipertrofie) (Figura 1). Nu au fost observate crampe, mialgii și ondulări. Examinările pneumologice complete (spirometrie și polisomnografie) și cardiologice (ecografie, monitorizare Holter ECG 24-h, RMN cardiac, inspecție fizică cardiologică completă cu istoric medical vizat și analiză reflexă cardiovasculară) nu au evidențiat nicio anomalie semnificativă. De asemenea, am exclus deficitul de maltază acidă (teste normale ale activității enzimatice în limfocite/leucocite). Imagistica prin rezonanță magnetică musculară (IRM) a arătat o implicare proeminentă a brâului scapular, a mușchilor paravertebrali, a mușchilor compartimentului posterior al coapsei (cu relativă economisire a mușchilor sartorius și gracilis) și a piciorului (în special gastrocnemius medialis și soleus) (Figura 2). Acest model de implicare a fost deja raportat în literatura de specialitate privind imaginea RMN în calpainopatie (19, 20). În plus, studiul RMN a arătat că mușchii gambei au fost efectiv atrofici și caracterizați prin infiltrarea grăsimilor, provocând o „mărire” musculară.”Prezentarea clinică, vârsta de debut, rata progresiei, distribuția slăbiciunii musculare și rezultatele RMN în proband au fost pe deplin în concordanță cu un diagnostic de LGMD2A descris pe larg în literatură (4, 21, 22).
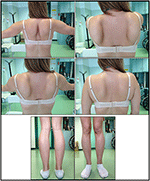
Figura 1. Proband caracteristici clinice: scapule înaripate și combinație de atrofie a coapsei și pseudo-hipertrofie a vițelului.
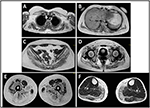
Figura 2. Proband musculare RMN: implicarea brâului membrelor superioare și a mușchilor paravertebrali (A, B), implicarea brâului membrelor inferioare și a compartimentului posterior al coapsei (C–E, mai ales glutei) cu relativă economisire a sartorius și gracilis și implicarea compartimentului posterior al piciorului (F, mai ales gastrocnemius medialis/soleus).
evaluarea membrilor familiei probandului nu a evidențiat niciun istoric de boală neuromusculară și nici o dovadă de consangvinitate între părinți. Cu toate acestea, mama probandului, la vârsta de 55 de ani, a prezentat 2 episoade sincopale pentru care a fost diagnosticată cu bradicardie idiopatică (până la 40 bpm noaptea). Investigații cardiologice extinse asupra mamei probandului au diagnosticat un bloc atrioventricular simptomatic (AV) după apariția mai multor episoade sincopale/lipotimie. Analiza a arătat prezența unui bloc AV de gradul I de bază, cu mai multe perioade de bradicardie severă, în cea mai mare parte nocturnă și adesea simptomatică, asociată cu unele faze ale blocului AV de gradul II, atât de tip 1, cât și 2, și unele faze ale disocierii AV complete nocturne. Ecocardiografia și testul ergometric nu au evidențiat modificări semnificative, cu excepția brevetului foramen ovale. Mama nu a prezentat niciun alt simptom clinic, condiție predispozantă sau factor de risc. Având în vedere imaginea clinică, mama probandului a fost implantată cu PMK. În plus, istoria familiei a arătat că bunica și străbunicul probandului au fost, de asemenea, implantate cu PMK la vârsta de 55 și, respectiv, 30 de ani. Mai mult, fratele bunicii probandului a murit la 51 de ani pentru cardiomiopatie dilatativă severă. Examinările cardiologice au fost, de asemenea, efectuate asupra tatălui și unchiului matern al probandului care a rezultat complet neafectat.
studiul de față a fost aprobat de Comitetul de etică al Fundației Santa Lucia și a fost realizat conform Declarației de la Helsinki. Toți participanții au oferit consimțământul informat semnat pentru analiza genetică și, în acest sens, au oferit și consimțământul pentru publicarea acestui raport de caz.
investigații de laborator și teste de Diagnostic
ADN-ul Genomic a fost extras din sângele periferic (400 unqql) folosind kitul de extracție a ADN-ului din sânge MagPurix și sistemul de extracție automată MagPurix (Resnova) conform instrucțiunilor producătorului. Probele au fost secvențiate folosind sistemul Ion PGM și panoul personalizat Ion Ampliseq specificitate ridicată (Thermo Fisher Scientific). Dimensiunea panoului a fost de 129.13 Kb, care este de așteptat să ecraneze ~99,72% din panoul total cu o acoperire minimă de 20X. Panoul a inclus 18 gene, care au fost selectate folosind literatura științifică, GeneReviews (www.ncbi.nlm.nih.gov/books/NBK1116/) și frecvența variantelor patogene în populația generală. O descriere detaliată a panoului NGS a fost rezumată în tabelul 1.
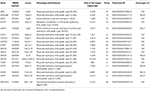
Tabelul 1. Panou NGS personalizat utilizat pentru diagnosticul LGMD.
construcția bibliotecilor a fost realizată de Ion AmpliSeq 3.0. Pentru reacțiile multiplex PCR s-au utilizat aproximativ 10 ng/ecqtl de ADN inițial. Succesiv, au fost efectuate două etape de purificare (folosind AMPure XP, Beckman Coulter) pentru a elimina contaminanții nedoriti și a fost efectuat un PCR final. Etapele de amplificare și îmbogățire a șabloanelor au fost realizate de Ion PGM Hi-Q OT2 kit-400, Ion OneTouch 2 System și Ion OneTouch ES (Thermo Fisher Scientific). Probele au fost prelucrate de Ion PGM Hi-Q Sequencing Kit (400 bp, Thermo Fisher Scientific) și rula pe Ion 316 cip v2 (850 fluxurile necesare) și Ion PGM Sequencer (Thermo Fisher Scientific). Rezultatele au fost analizate folosind Ion Reporter 4.6 (Thermo Fisher Scientific) și Integrated Genome Viewer (IGV). Interpretarea variantelor genetice a fost realizată de Human Gene Mutation Database (HGMD), Leiden Open Variation Database (LOVD), ClinVar și ExAC. Efectul funcțional al variantelor detectate a fost evaluat prin instrumente predictive bioinformatice, inclusiv Mutation Taster, Varsome, SIFT, PolyPhen 2, SMART, Human Splicing Finder (HSF). Secvențiere directă (Bigdye Terminator v3.1, Terminator BigDyeX și ABI3130, Biosystems aplicate) a fost efectuată pentru a confirma variantele genetice și pentru a secvențializa regiunile de codificare genomică cu o acoperire <20X (LMNA, Chr1:156106052-156106076; DYSF, Chr2:71753352-71753502, Chr2:71776385-71776640; Sgcb, Chr4:52904225-52904560; sgca, chr17:48243242-48243570).
rezultate
analiza NGS a probandului a evidențiat 3 variante heterozigote (figura suplimentară 1). Două variante au fost localizate în CAPN3, și anume NM_000070.2 (CAPN3): c.550dela (P.Thr184Argfs) și c.1813g>C (P.Val605Leu) în exonii 4 și respectiv 16. Prima este o singură deleție nucleotidică și este cea mai frecventă variantă patogenă (75% din cazuri) în țările europene (1). Așa cum era de așteptat, analiza bioinformatică a clasificat c.550dela (rs80338800) ca o variantă de pierdere a funcției (P.Thr184Argfs), provocând o schimbare a cadrului cadrului de citire deschis. Instrumentele predictive (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) au descris varianta ca fiind dăunătoare. În plus, SMART a dezvăluit că produsul proteic modificat nu are domeniul calpain 3 și cele trei motive „EF-hands”. Primul domeniu este implicat în căile de semnalizare ale Calpain, în timp ce mâinile EF sunt esențiale pentru activarea dependentă de Ca++a proteinei (4).
referitor la c.1813g>C, este o variantă nouă de missense (P.Val605Leu) care a fost prezisă a fi dăunătoare (prin mutație degustător, HSF, Varsome, polifen 2, cernere) din cauza unei potențiale modificări a îmbinării. Această variantă nu este adnotată în literatură sau în bazele de date online și nu a fost găsită la 200 de subiecți de control. Din păcate, analiza de către SMART tool nu a dat rezultate semnificative, deoarece varianta se află într-un domeniu necaracterizat. Analiza segregării CAPN3 a arătat că mama și unchiul matern erau ambii heterozigoți pentru c.550dela, în timp ce tatăl era purtător de c.1813g>C.
în plus, analiza NGS asupra probandului a relevat prezența unei variante noi în LMNA, și anume NM_170707 (LMNA): c.550c>T (P.Gln184*). Varianta supra-menționată a fost prezisă a fi o variantă nulă, nu a fost încă descrisă în literatură sau în bazele de date online și nu a fost găsită la 200 de subiecți de control. Instrumentele Predictive (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) au descris un efect patogen al acestei variante asupra produsului proteic. Instrumentul inteligent a raportat că proteina lmna modificată nu are domeniul filamentului, care este esențial pentru a-și menține structura și funcția. Analiza segregării a evidențiat prezența c.550c > varianta T numai la mamă, sugerând o posibilă asociere cu simptomatologia cardiacă și istoricul familial al bolii.
conform criteriilor stabilite de Colegiul American de Genetică Medicală (ACMG) standarde și orientări (23), C.1813g>C (CAPN3) poate fi descrisă ca o variantă patogenă probabilă, având în vedere că este localizată într-un domeniu critic fără variație benignă (PM1); este absent în bazele de date ExAc, GnomAD și 1000 Genome Browser (PM2); având în vedere modelul recesiv de moștenire, acesta a fost detectat în trans cu o variantă patogenă (PM3); mai multe linii de dovezi computaționale susțin un efect dăunător asupra genei sau produsului genetic (PP3). În ceea ce privește clasificarea clinică a c. 550c>T (LMNA) de către ACMG, aceasta poate fi desemnată ca variantă patogenă, deoarece este o variantă nulă care provoacă pierderea funcției (P.Gln184*) în LMNA (PVS1); este absent în bazele de date ExAc, GnomAD și 1000 Genome Browser (PM2); este localizat într-un domeniu critic fără variație benignă (PM1); mai multe linii de dovezi computaționale susțin un efect dăunător asupra genei sau produsului genetic (PP3).
concluzii
acest raport de caz a prezentat un pacient afectat de LGMD, care s-a dovedit a fi purtător de mutații nu numai în CAPN3 (c.550dela și c.1813g>C), ci și în LMNA (c.550c > T). Aceste rezultate explică fenotipul neuromuscular al probandului din cauza mutațiilor CAPN3 și evidențiază un risc potențial de tulburări cardiovasculare datorită prezenței variantei în LMNA și a istoricului familial pozitiv. Având în vedere aceste rezultate, membrii familiei au fost supuși analizei segregării. În special, mama a rezultat să fie purtătoare de CAPN3_c.550dela și, respectiv, LMNA_c.550c>T. Mama poate fi considerată un purtător sănătos pentru mutația patogenă CAPN3_c.550dela asociată cu calpainopatia. Cu toate acestea, prezența unei variante noi în LMNA poate explica patologia cardiovasculară (bradicardie și episoade sincopale). Absența simptomatologiei neuromusculare la mamă și imaginea clinică particulară a probandului au exclus o posibilă asociere a LMNA_c.550c>T cu fenotip neuromuscular, în special în ceea ce privește distrofia musculară Emery-Dreifuss (EMD). De fapt, EMD afectează în mod specific mușchii vastus lateralis și biceps brachii, care sunt relativ cruțați în LGM2A (24, 25).
unchiul matern a fost heterozigot doar pentru CAPN3_c.550dela și, așa cum era de așteptat, nu a prezentat probleme neuromusculare sau cardiovasculare. Tatăl a dus la purtarea romanului CAPN3_c.1813g > C și, prin urmare, nu a fost afectat. În ansamblu, analiza segregării a confirmat moștenirea celor trei mutații ale probandului de la rudele sale și a evidențiat o familiaritate pentru cardiomiopatie care nu poate fi neglijată (Figura 3).
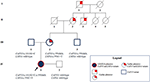
Figura 3. Pedigree care arată familiaritatea pozitivă pentru fenotipul cardiac moștenit de descendența maternă și transmiterea variantelor CAPN3 și LMNA la membrii familiei.
în total, aceste date ridică câteva considerații importante. În primul rând, panoul NGS la acest pacient a fost esențial pentru a ajunge la diagnosticul molecular al calpainopatiei, detectând o mutație cunoscută și o a doua variantă nouă prezisă ca patogenă. În al doilea rând, panoul NGS a permis identificarea noii variante LMNA_C.550c>T în proband. Acest rezultat a fost în concordanță cu istoricul familial pozitiv și cu datele de segregare, explicând bolile cardiovasculare la mamă și, mai important, recomandând o urmărire cardiologică mai specifică în proband. Este important de menționat că manifestările cardiologice au apărut la mamă mai târziu în Vârstă (55 de ani), astfel încât să putem presupune că probandul ar putea fi încă într-o „fază cardiologică asimptomatică.”Cu toate acestea, o expresie fenotipică variabilă a mutației LMNA în proband comparativ cu mama nu poate fi exclusă. Toate aceste date subliniază importanța unei abordări integrate între clinicieni și geneticieni, pentru interpretarea corectă a rezultatelor, consilierea genetică adecvată și, eventual, managementul clinic și urmărirea. În ceea ce privește consilierea genetică și familială, analiza NGS a fost efectuată și pe partenerul probandului, pentru a estima riscul de reproducere pentru cuplu. Partenerul a fost negativ la toate cele 18 gene testate, ceea ce înseamnă că are un risc rezidual de 1/650 pentru a fi un purtător sănătos pentru mutațiile cauzale ale LGMD, având în vedere că testul este sensibil la 84%. Luând în considerare profilul genetic al probandului și sensibilitatea testului NGS, riscul rezidual pentru cuplu de a avea un copil afectat de calpainopatie este de 1/1300. Pe de altă parte, variantele patogene LMNA sunt transmise după un model dominant autosomal, ceea ce înseamnă că probandul are o probabilitate de 50% de a avea copii heterozigoți. Cu toate acestea, imaginea clinică a descendenților nu poate fi cu siguranță prezisă, deoarece LMNA_c.550c>T este o variantă nouă și impactul său funcțional asupra fenotipului este încă necunoscut.
în concluzie, acest raport de caz evidențiază utilitatea clinică a panourilor NGS pentru a oferi diagnostic precis LGMD2A și descrie fenotipuri complexe și comorbidități provenite din moștenirea diferitelor mutații în gene multiple. Cu toate acestea, aplicarea NGS în practica clinică ar trebui să fie întotdeauna combinată cu o consiliere pre – și post-genetică pentru a oferi o explicație clară a rezultatelor, posibilele implicații asupra fenotipului pacienților, riscul de recurență în cadrul familiei, precum și pentru a explica posibile constatări neașteptate.
disponibilitatea datelor
nu au fost generate sau analizate seturi de date pentru acest studiu.
declarație de etică
acest studiu a fost realizat în conformitate cu recomandările Comitetului de etică al Fundației Santa Lucia, cu acordul scris informat al tuturor subiecților. Toți subiecții și-au dat consimțământul scris în cunoștință de cauză în conformitate cu declarația de la Helsinki. Protocolul a fost aprobat de Comitetul de etică al Fundației Santa Lucia.
contribuții ale autorului
RC, CSt, VC, GC, RG, GPa, SC, CP și JM au contribuit la achiziționarea de date, analiza și interpretarea datelor. CSt, RC, GC, RG, GM și EG au fost implicate în redactarea manuscrisului. SZ, GPr, CSa și SS au fost implicate în achiziționarea de date clinice. RC, CSt, VC, GC, RG, GPa, SC, CP, JM, SZ, GPr, GM, CSA, SS și EG au dat aprobarea finală a versiunii care urmează să fie publicată.
finanțare
această lucrare este susținută de 5x 2016 Ministerul Național al Sănătății 2.
Declarație privind conflictul de interese
autorii declară că cercetarea a fost realizată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
material suplimentar
materialul suplimentar pentru acest articol poate fi găsit online la: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. proteine si diagnostic genetic al membrelor brâu distrofie musculară de tip 2A: randamentul și capcanele. Nervul Muscular. (2015) 52:163–73. doi: 10.1002 / mus.24682
Rezumat PubMed / CrossRef Text Complet / Google Scholar
2. Nigro V, Savarese M. baza genetică a distrofiilor musculare ale membrelor-brâu: actualizarea din 2014. Acta Myol. (2014) 33:1–12.
Rezumat PubMed / Google Scholar
3. Richard I, Hogrel JY, Stockholm D, Payan CAM, Fougerousse F, Eymard B, și colab. Istoricul natural al LGMD2A pentru delimitarea măsurilor de rezultat în studiile clinice. Ann Clin Transl Neurol. (2016) 3:248–65. doi: 10.1002 / acn3.287
Rezumat PubMed / CrossRef Text Complet / Google Scholar
4. Angelini C, Fanin M. Calpainopatie. În: Adam MP, Ardinger HH, Pagon ra editori. GeneReviews-ul. Seattle, WA: Universitatea din Washington (2017) 1-30.
Rezumat PubMed / Google Scholar
5. Kyriakides T, Angelini C, Schaefer J, Sacconi S, Siciliano G, Vilchez JJ, și colab. Ghidurile EFNS privind abordarea diagnostică a hipercemiei PAUCI-sau asimptomatice. Eur J Neurol. (2010) 17:767–73. doi: 10.1111 / j. 1468-1331.2010. 03012.x
CrossRef Text Complet / Google Scholar
6. Mori-Yoshimura M, Segawa K, Minami N, Oya Y, Komaki H, Nonaka I și colab. Disfuncție cardiopulmonară la pacienții cu distrofie musculară la nivelul membrelor 2A. nervul muscular. (2017) 55:465–9. doi: 10.1002 / mus.25369
CrossRef Text Complet / Google Scholar
7. Okere a, Reddy SS, Gupta s, Shinnar M. o cardiomiopatie la un pacient cu distrofie musculară a brâului membrelor de tip 2a. (2013) 6:e12–13. doi: 10.1161 / CIRCHEARTFAILURE.112.971424
CrossRef Text Complet / Google Scholar
8. Kramerova I, Ermolova N, Eskin A, Hevener A, Quehenberger O, Armando AM și colab. Nerespectarea transcripției genelor necesare adaptării musculare stă la baza distrofiei musculare a brâului membrelor 2A (calpainopatie). Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093 / hmg / ddw086
PubMed rezumat / CrossRef Text Complet / Google Scholar
9. Thompson R, Straub V. distrofii musculare ale brâului membrelor – colaborări internaționale pentru cercetarea translațională. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038 / nrneurol.2016.35
Rezumat PubMed / CrossRef Text Complet / Google Scholar
10. Strafella C, Caputo V, Galota MR, Zampatti S, Marella G, Mauriello S, și colab. Aplicarea medicamentelor de precizie în bolile neurodegenerative. Neurol Frontal. (2018) 9:701. doi: 10.3389 / fneur.2018.00701
Rezumat PubMed / CrossRef Text Complet / Google Scholar
11. Cascella R, Strafella C, Caputo V, Errichiello V, Zampatti s, Milano F, și colab. Spre aplicarea medicamentelor de precizie în degenerescența maculară legată de vârstă. Prog Retin Eye Res. (2017) 63:132-46. doi: 10.1016/j.preteyeres.2017.11.004
Rezumat PubMed / CrossRef Text Complet / Google Scholar
12. Cascella R, Strafella C, Longo G, Manzo L, Ragazzo M, de Felici C, și colab. Evaluarea riscului individual pentru AMD cu consiliere genetică, istoric familial și testare genetică. Ochi. (2017) 32:446–50. doi: 10.1038/ochi.2017.192
Rezumat PubMed / CrossRef Text Complet / Google Scholar
13. Adams DR, Ing CM. Secvențiere de generație următoare pentru a diagnostica tulburările genetice suspectate. N Engl J Med. (2019) 379:1353–62. doi: 10.1056 / NEJMc1814955
PubMed rezumat / CrossRef text integral / Google Scholar
14. Angelini C. Boala neuromusculară. Diagnostic și descoperire în distrofia musculară a membrelor. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038 / nrneurol.2015.230
CrossRef Text Complet / Google Scholar
15. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, și colab. Utilizarea secvențierii întregului exom pentru diagnosticarea distrofiei musculare a membrelor: rezultate și lecții învățate. JAMA Neurol. (2015) 72:1424–32. doi: 10.1001 / jamaneurol.2015.2274
Rezumat PubMed / CrossRef Text Complet / Google Scholar
16. Milic A, Daniele N, Lochm Oktiller H, Mora M, Comi GP, Moggio M, și colab. O treime din biopsiile LGMD2A au activitate proteolitică calpain 3 normală, determinată printr-un test in vitro. Tulburări Neuromusc. (2007) 17:148–56. doi: 10.1016 / j.nmd.2006.11.001
Rezumat PubMed / CrossRef Text Integral / Google Scholar
17. Lanzillo R, Aurino S, Fanin M, Aguennoz M, Vitale F, Fiorillo C, și colab. Calpainopatie cu debut precoce cu nivel normal de calpain nefuncțional 3. Dev Med Neurol Copil. (2006) 48:304–6. doi: 10.1017 / S001216220600065x
PubMed rezumat | CrossRef text integral / Google Scholar
18. Talim B, Ognibene A, Mattioli e, Richard I, Anderson LV, Merlini L. expresia calpain normală în distrofia musculară confirmată genetic de tip 2a. Neurologie. (2001) 56:692–3. doi: 10.1212 / wnl.56.5.692-a
CrossRef Text Complet / Google Scholar
19. D-manera J, Llauger J, Gallardo e, Illa I. RMN muscular în distrofii musculare. Acta Myol. (2015) 34:95–108.
Rezumat PubMed / Google Scholar
20. Mercuri e, Bushby K, Ricci E, Birchall DR, Pane M, Kinali M și colab. Rezultatele RMN musculare la pacienții cu distrofie musculară a brâului membrelor cu deficit de calpain 3 (LGMD2A) și contracturi timpurii. Tulburări Neuromusc. (2005) 15:164–71. doi: 10.1016 / j.nmd.2004.10.008
PubMed Rezumat / CrossRef Text Integral / Google Scholar
21. Magri F, Nigro V, Angelini C, Mongini t, Mora M, Moroni i, și colab. Registrul italian de distrofie musculară a brâului membrelor: frecvența relativă, caracteristicile clinice și diagnosticul diferențial. Nervul Muscular. (2017) 55:55–68. doi: 10.1002 / mus.25192
Rezumat PubMed / CrossRef Text Complet / Google Scholar
22. Chae J, Minami N, Jin Y, Nakagawa M, Murayama K, Igarashi F, și colab. Calpain 3 mutații genetice: constatări genetice și clinico-patologice în distrofia musculară a membrelor. Tulburări Neuromusc. (2001) 11:547–55. doi: 10.1016 / S0960-8966(01)00197-3
PubMed rezumat / CrossRef Text Complet / Google Scholar
23. Richards S, Aziz N, Bale s, Bick D, Das s, Gastier-Foster J, și colab. Standarde și orientări pentru interpretarea variantelor de secvență: o recomandare comună de consens a Colegiului American de Genetică Medicală și genomică și a Asociației pentru Patologie Moleculară. Genet Med. (2015) 17:405–24. doi: 10.1038 / gim.2015.30
Rezumat PubMed / CrossRef Text Complet / Google Scholar
24. Bonne G, Mercuri E, Muchir A, Urtizberea A, B Centocane Hm, Recan D și colab. Spectrul genetic clinic și molecular al distrofiei musculare autozomale dominante Emery-Dreifuss datorită mutațiilor genei lamin A/C. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170::aid-ANA6>3.0.CO; 2-J
rezumat PubMed / CrossRef Text Complet / Google Scholar
25. Hong JS, Ki CS, Kim JW, Suh il, Kim JS, Baek KK și colab. Disritmii cardiace, cardiomiopatie și distrofie musculară la pacienții cu distrofie musculară Emery-Dreifuss și distrofie musculară de tip 1B. J coreeană med Sci. (2005) 20:283–90. doi: 10.3346 / jkms.2005.20.2.283
CrossRef Full Text / Google Scholar