- Introducere
- metode
- filtrarea datelor de profilare ribozomală
- asamblarea Genomurilor virtuale
- determinarea regiunii Ribo-seq Read-mapate pe o joncțiune (RMRJ) de circRNAs
- instruirea Modelului și clasificarea RMRJs
- Predicția peptidelor traduse de RMRJs
- rezultate și discuții
- circRNAs traduse la om și A. thaliana
- îmbogățirea funcțională a circrn-urilor umane și A. thaliana cu potențial de codificare
- test de precizie pentru CircCode
- influența adâncimii de secvențiere a datelor Ribo-seq
- Compararea CircCode cu alte instrumente
- concluzii
- disponibilitate și cerințe
- declarație de disponibilitate a datelor
- contribuții autor
- finanțare
- Conflict de interese
- material suplimentar
Introducere
ARN-urile circulare (circrna) sunt un tip special de moleculă de ARN necodificatoare care a devenit un subiect fierbinte de cercetare în domeniul ARN și primește o mare atenție (Chen și Yang, 2015). Comparativ cu ARN-urile liniare tradiționale (care conțin capete de 5′ și 3′), moleculele de arnc au de obicei o structură circulară închisă; făcându-le mai stabile și mai puțin predispuse la degradare (Vicens și Westhof, 2014). Deși existența circrna este cunoscută de ceva timp, aceste molecule au fost considerate a fi un produs secundar al îmbinării ARN. Cu toate acestea, odată cu dezvoltarea tehnologiilor de secvențiere și bioinformatică cu randament ridicat, circrn-urile au devenit recunoscute pe scară largă la animale și plante (Chen și Yang, 2015). Studii recente au arătat, de asemenea, că un număr mare de circRNAs pot fi traduse în peptide mici în celule (Pamudurti și colab., 2017) și au roluri cheie în ciuda nivelului lor uneori scăzut de exprimare (Hsu și Benfey, 2018; Yang și colab., 2018). Deși se identifică un număr tot mai mare de arnc, funcțiile lor la plante și animale rămân, în general, de studiat. În plus față de funcțiile lor ca momeli miRNA, circrna-urile au un potențial de translație important, dar nu sunt disponibile instrumente pentru prezicerea specifică a capacităților de translație ale acestor molecule (Jakobi și Dieterich, 2019).
există mai multe instrumente pentru predicția și identificarea circrna-urilor, cum ar fi Ciri (Gao și colab., 2015), CIRCexplorer (Dong și colab., 2019), CircPro (Meng și colab., 2017) și circtools (Jakobi și colab., 2018). Printre acestea, CircPro poate dezvălui circRNAs traduse prin calcularea unui scor potențial de traducere pentru circRNAs bazat pe CPC (Kong și colab., 2007), care este un instrument pentru identificarea cadrului de citire deschis (ORF) într-o secvență dată. Cu toate acestea, deoarece unele circrna nu folosesc codonul de pornire în timpul traducerii (Ingolia și colab., 2011; Slavoff și colab., 2013; Kearse și Wilusz, 2017; Spealman și colab., 2018), angajarea CPC poate filtra unele circRNAs cu adevărat traduse. În acest studiu, am folosit BASiNET (Ito și colab., 2018), care este un clasificator ARN bazat pe metodele de învățare automată (random forest și modelul J48). Inițial transformă ARN-urile de codare date (date pozitive) și ARN-urile necodificate (date negative) și le reprezintă ca rețele complexe; apoi extrage măsurile topologice ale acestor rețele și construiește un vector de caracteristici pentru a instrui modelul care este utilizat pentru a clasifica capacitatea de codificare a circrn-urilor. Cu această metodă, se evită filtrarea eronată a circRNAs traduse care nu sunt inițiate de AUG. În plus, tehnologia Ribo-seq, care se bazează pe secvențierea cu randament ridicat pentru a monitoriza RPF-urile (fragmente protejate ribozomal) ale transcrierilor (Guttman și colab., 2013; Brar și Weissman, 2015), pot fi utilizate pentru a determina locațiile circRNAs care sunt traduse (Michel și Baranov, 2013). Pentru a identifica capacitatea de codificare a circrna–urilor, am dezvoltat instrumentul CircCode, care implică un cadru bazat pe Python 3 și am aplicat CircCode pentru a investiga potențialul de traducere al circrna-urilor de la oameni și Arabidopsis thaliana. Munca noastră oferă o resursă bogată pentru studierea în continuare a funcțiilor circRNAs cu capacitate de codificare.
metode
CircCode a fost scris în limbajul de programare Python 3; folosește Trimmomatic (Bolger și colab., 2014), bowtie (Langmead și Salzberg, 2012) și STAR (Dobin și colab., 2013) pentru a filtra Raw Ribo-seq citește și mapează aceste citiri filtrate la genom. CircCode identifică apoi Ribo-seq regiuni mapate în circRNAs care conțin joncțiuni. După aceea, secvențele mapate candidate din circRNAs sunt sortate pe baza clasificatorilor (modelul J48) în ARN-uri de codificare și ARN-uri necodificate de BASiNET. În cele din urmă, peptidele scurte produse prin traducere sunt identificate ca potențiale regiuni de codificare a circrna-urilor. Întregul proces de CircCode este format din cinci pași (Figura 1).
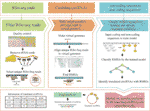
Figura 1 fluxul de lucru al CircCode. Stratul superior reprezintă fișierul de intrare Necesar pentru fiecare pas al CircCode. Stratul de mijloc este împărțit în trei părți și fiecare parte reprezintă o etapă diferită de funcționare. De la stânga la dreapta, prima parte reprezintă filtrarea datelor Ribo-seq; controlul calității este executat prin Trimmomatic, iar citirile rRNA sunt îndepărtate prin bowtie. A doua parte reprezintă pașii utilizați pentru producerea genomului virtual și alinierea citirilor filtrate la genomul virtual cu stea. Ultima parte reprezintă identificarea circrna-urilor traduse prin învățarea automată. Stratul inferior reprezintă ultima etapă utilizată pentru a prezice peptidele traduse din circRNAs și rezultatele finale de ieșire, inclusiv informații despre circRNAs traduse și produsele lor de traducere.
filtrarea datelor de profilare ribozomală
în primul rând, fragmentele și adaptoarele de calitate scăzută din citirile Ribo-Seq sunt eliminate prin Trimmomatic cu parametrii impliciți pentru a obține citiri Ribo-seq curate. În al doilea rând, aceste citiri Ribo-seq curate sunt mapate la o bibliotecă rRNA pentru a elimina citirile derivate din rRNA folosind bowtie. Deoarece lungimile de citire ale Ribo-seq sunt relativ scurte (în general mai mici de 50 bp), este posibil ca o citire să se potrivească mai multor regiuni. În acest caz, este dificil să se determine ce regiune corespunde unei anumite citiri. Pentru a evita acest lucru, citirile Ribo-seq curate sunt mapate la genomul unei specii de interes, iar citirile care nu sunt perfect aliniate la genom sunt considerate ultimele citiri Ribo-seq unice.
asamblarea Genomurilor virtuale
Circrna-urile apar de obicei ca molecule în formă de inel în eucariote și pot fi identificate pe baza joncțiunilor lor de îmbinare din spate. Cu toate acestea, secvențele de circrna din fișierul fasta sunt adesea în formă liniară. În teorie, rezultatul indică faptul că joncțiunea se află între nucleotida terminală 5′ și nucleotida terminală 3′, deși joncțiunea și secvența din apropierea joncțiunii nu pot fi vizualizate direct, aliniind astfel citirile Ribo-seq la secvențele circRNA, inclusiv joncțiunile, într-o manieră directă.
CircCode conectează secvența fiecărui circRNA în tandem astfel încât joncțiunea pentru fiecare să fie în mijlocul secvenței nou construite. De asemenea, am separat fiecare unitate de serie cu 100 N nucleotide pentru a evita confuzia la etapa de aliniere a secvenței (lungimea fiecărui RPF este mai mică de 50 bp). În cele din urmă, am obținut un genom virtual format doar din circrna candidate în tandem separate de 100 Ns. Deoarece CircCode se concentrează doar pe alinierea dintre citirile Ribo-seq și secvențele circRNA, putem investiga potențialul de codificare al circrna prin cartografierea citirilor Ribo-seq la acest genom virtual, care poate economisi o cantitate mare de timp de calcul (genomul virtual este mult mai mic decât întregul genom) și crește precizia (prin evitarea interferențelor între comparațiile secvențelor din amonte și din aval ale circrna).
determinarea regiunii Ribo-seq Read-mapate pe o joncțiune (RMRJ) de circRNAs
citirile finale unice Ribo-seq sunt mapate la un genom virtual creat anterior folosind STAR. Deoarece fiecare unitate circRNA tandem a fost separată de 100 n baze înainte de a produce genomul virtual, cea mai mare lungime de intron a fost setată să nu depășească 10 Baze cu parametrul „–alignIntronMax 10.”Acest parametru elimină orice interacțiune între diferite circRNAs în alinierea secvenței. În a doua etapă a producției de genom virtual, CircCode stochează informații de joncțiune pozițională pentru fiecare circRNA din genomul virtual. Dacă Regiunea cartată citită Ribo-seq din genomul virtual include joncțiunea circRNA, iar numărul de citiri Ribo-seq mapate pe joncțiune (NMJ) este mai mare de 3, Regiunea cartată Ribo-seq pe joncțiunea circrna poate fi privită ca un RMRJ, care dezvăluie un segment aproximativ tradus de circrna în apropierea locului de joncțiune.
instruirea Modelului și clasificarea RMRJs
deși RMRJs poate constitui o dovadă puternică a traducerii, există încă unele deficiențe în această metodă. Deoarece lungimea citirilor hărții ribozomale este scurtă, o citire poate fi comparată cu poziția greșită. Prin urmare, nu este convingător să se considere pur și simplu Regiunea acoperită de citirile Ribo-seq drept regiunea tradusă. În acest scop, metoda de învățare automată este utilizată pentru a identifica capacitatea de codificare a RMRJ. În primul rând, CircCode extrage ARN-uri de codificare (date pozitive) și ARN-uri necodificate (date negative) dintr-o specie de interes și le folosește pentru formarea modelului prin intermediul diferenței de vectori de caracteristici între ARN-uri de codificare și necodificare. CircCode folosește apoi modelul instruit pentru a clasifica Rmrj-urile obținute în pasul anterior de BASiNET. Dacă RMRJ al unui circRNA este recunoscut ca ARN codificator, atunci acest circRNA poate fi identificat ca un circRNA tradus.
Predicția peptidelor traduse de RMRJs
deoarece expresia circrna în organisme este scăzută, datele Ribo-seq nu arată în mod clar periodicitatea exactă a 3-nt în cazul unui număr mai mic de RPF. Prin urmare, este dificil să se determine locul exact de pornire a traducerii unui circRNA tradus. Datorită prezenței unui codon stop în unele Rmrj și deoarece codonul start este dificil de determinat, metoda de a găsi un ORF bazat pe un codon start și un codon stop nu este fezabilă.
pentru a determina regiunile de traducere adevărate ale acestor circrna și pentru a genera produsul final de traducere, FragGeneScan (Rho și colab., 2010), care poate prezice regiunile de codificare a proteinelor în gene fragmentate și gene cu schimbări de cadre, este utilizat pentru a determina peptidele traduse produse de circRNAs.
pentru a evita procesul de rulare greoaie, toate modelele pot fi numite de un script shell; utilizatorul poate pur și simplu să completeze fișierul de configurare dat și să îl introducă în script, iar întregul proces de predicție a circRNAs traduse va fi apoi rulat. În plus, CircCode poate fi rulat separat, pas cu pas, astfel încât utilizatorul să poată ajusta parametrii în mijlocul procedurii și să vizualizeze rezultatele fiecărui pas după dorință.
rezultate și discuții
după testarea pe mai multe computere, CircCode a fost găsit pentru a rula cu succes cu dependențele necesare instalate. Pentru a testa performanța CircCode, am folosit date pentru oameni și A. thaliana pentru a prezice circRNAs cu potențial de traducere. Rezultatele au fost comparate cu arnc care au fost verificate experimental ca confirmare. Ulterior, am testat valoarea false discovery rate (FDR) a CircCode în continuare. Am folosit GenRGenS (Ponty și colab., 2006) pentru a genera un set de date pentru testare pe baza circrna-urilor traduse cunoscute și a confirmat că valoarea FDR se încadrează într-un interval acceptabil și la un nivel scăzut. În cele din urmă, am evaluat efectul diferitelor adâncimi de secvențiere a datelor Ribo-seq asupra predicțiilor CircCode și am comparat CircCode cu alte programe software.
circRNAs traduse la om și A. thaliana
pentru a aplica instrumentul CircCode datelor reale, am descărcat mai întâi fișierele, inclusiv genomul de referință uman GRCh38, adnotarea genomului și ARNr uman, de la Ensembl. Pentru A. thaliana, genomii de referință (TAIR10), fișierele de adnotare a genomului și secvențele rRNA corespunzătoare au fost toate descărcate de la plantele Ensembl. Datele Ribo – seq pentru oameni și A. thaliana au fost descărcate de pe RPFdb (numere de aderare: GSE96643, GSE81295, GSE88794) (Hsu și colab., 2016; Willems și colab., 2017) și toate circrn-urile candidate de la human și A. thaliana au fost descărcate de pe CIRCPedia v2 (Dong și colab., 2018) și, respectiv, PlantcircBase (Chu și colab., 2017). În cele din urmă, am identificat 3.610 circRNAs traduse de la om și 1.569 circRNAs traduse de la A. thaliana folosind CircCode (date suplimentare 1).
îmbogățirea funcțională a circrn-urilor umane și A. thaliana cu potențial de codificare
folosind rezultatele CircCode pentru human și A. thaliana, instrumentul online KOBAS 3.0 (Wu și colab., 2006) a fost angajat pentru a adnota aceste circrna traduse pe baza genelor lor părinte. Mai mult, am efectuat analiza funcțională GO (Gene Ontology) și analiza de îmbogățire KEGG (Kyoto Encyclopedia of Genes and Genomes) pentru aceste circRNAs traduse folosind pachetul r clusterProfiler (Yu și colab., 2012).
rezultatele KEGG au arătat că circrn-urile umane au fost îmbogățite în procesarea proteinelor în calea reticulului endoplasmatic, calea metabolismului carbonului și calea de transport a ARN-ului. Analiza GO a indicat participarea circrna-urilor traduse umane la reglarea legării moleculelor, a activității ATPazei și a altor procese biologice legate de îmbinarea ARN. În plus, circuitele traduse ale lui A. thaliana sunt îmbogățite în căi legate de rezistența la stres, sugerând că acestea joacă roluri vitale în acest proces (date suplimentare 2).
test de precizie pentru CircCode
pentru a investiga acuratețea CircCode, au fost utilizate secvențe de testare generate de GenRGenS, care utilizează modelul Markov ascuns pentru a produce secvențe care au aceleași caracteristici de secvență (cum ar fi frecvențele diferitelor nucleotide, codoni diferiți și nucleotide diferite la începutul secvenței).
pentru acest studiu, am folosit circRNAs traduse umane publicate anterior (Yang și colab., 2017) ca intrare pentru GenRGenS și a generat 10.000 de secvențe pentru a testa CircCode. Am repetat testul de 10 ori și, în medie, 27 de circRNAs traduse au fost prezise de fiecare dată. Valoarea FDR a fost calculată la 0,0027, care este mult mai mică decât 0,05, indicând faptul că rezultatele prezise sunt credibile.
în plus, am comparat circrna-urile traduse de la oameni identificate prin CircCode cu datele verificate ale circRNA asociate polizomului (Yang și colab., 2017). Dintre acestea, 60% din arnc au fost identificate prin CircCode (date suplimentare 3).
influența adâncimii de secvențiere a datelor Ribo-seq
pentru a investiga impactul adâncimii de secvențiere a datelor Ribo-Seq asupra rezultatelor identificării CircCode, am testat mai întâi efectul adâncimii de secvențiere asupra numărului de circrna traduse (figura 2a). Când adâncimea de secvențiere a fost scăzută, numărul prezis de circRNAs traduse a fost scăzut, iar numărul de circRNAs traduse a crescut odată cu creșterea adâncimii de secvențiere. Numărul de circrn-uri traduse a devenit stabil atunci când adâncimea de secvențiere a atins nu mai puțin de 10% acoperire de transcriere liniară.

Figura 2 (a) efectul adâncimii de secvențiere a datelor Ribo-seq asupra numărului prezis de circrna traduse. (B) efectul numărului de citire a joncțiunii (JRN) asupra sensibilității CircCode la diferite adâncimi de secvențiere.
în al doilea rând, a fost evaluată și influența NMJ asupra sensibilității la diferite adâncimi de secvențiere (figura 2b). Rezultatele au arătat că NMJ a avut un impact mai mic asupra sensibilității pe măsură ce adâncimea de secvențiere a crescut. CircCode a avut, de asemenea, o sensibilitate mai mare atunci când se utilizează date Ribo-seq cu adâncime de secvențiere mai mare.
Compararea CircCode cu alte instrumente
pentru a compara CircCode cu alte instrumente, cum ar fi CircPro, același set de date Ribo-seq (SRR3495999) de la A. thaliana a fost folosit pentru a identifica circRNAs traduse folosind șase procesoare, cu 16 gigabytes de RAM. CircPro a identificat 44 de arnc traduse în 13 minute, în timp ce CircCode a identificat 76 de arnc traduse în 20 de minute. Astfel, CircCode este mai sensibil decât CircPro la același nivel hardware al computerului, dar durează mai mult timp. CircPro este concis și consumă mai puțin timp decât CircCode, dar CircCode poate identifica mai multe circRNAs cu capacitate de codificare decât CircPro.
concluzii
Arnc-urile joacă un rol important în biologie și este esențial să se identifice cu exactitate arnc-urile cu capacitatea de codificare pentru cercetările ulterioare. Bazat pe Python 3, am dezvoltat CircCode, un instrument de linie de comandă ușor de utilizat, care are o sensibilitate ridicată pentru identificarea circRNAs traduse din Ribo-Seq citește cu mare precizie. CircCode prezintă performanțe bune atât la plante, cât și la animale. Lucrările viitoare vor adăuga analiza caracterelor din aval la CircCode vizualizând fiecare pas al procesului și optimizând acuratețea predicției.
disponibilitate și cerințe
CircCode este disponibil la https://github.com/PSSUN/CircCode; sistem(e) de operare: Linux, limbaje de programare: Python 3 și R; alte cerințe: bedtools (versiunea 2.20.0 sau mai târziu), bowtie, STAR, Python 3 pachete (Biopython, Panda, rpy2), r-pachete (BASiNET, Biostrings). Pachetele de instalare pentru toate software-ul necesar sunt disponibile pe pagina de pornire CircCode. Utilizatorii nu trebuie să le descarce individual. Pagina de pornire CircCode oferă, de asemenea, manuale detaliate de utilizare pentru referință. Instrumentul este disponibil gratuit. Nu există restricții privind utilizarea de către nonacademics.
declarație de disponibilitate a datelor
toate datele relevante se află în manuscris și în fișierele sale de informații justificative.
contribuții autor
conceptualizare: PS, GL. Curarea datelor: PS, GL. Analiza formală: PS, GL. Redactare-Draft Original: PS, GL. Scriere-revizuire și editare: PS, GL.
finanțare
această lucrare a fost susținută de granturi de la Fundația Națională de științe Naturale din China (grant nos. 31770333, 31370329, și 11631012), programul pentru New Century excellent Talents in University (NCET-12-0896), și fondurile de cercetare fundamentală pentru universitățile Centrale (nr. GK201403004). Agențiile de finanțare nu au avut niciun rol în studiu, proiectarea acestuia, colectarea și analiza datelor, decizia de publicare sau pregătirea manuscrisului. Finanțatorii nu au avut niciun rol în proiectarea studiului, colectarea și analiza datelor, decizia de publicare sau pregătirea manuscrisului.
Conflict de interese
autorii declară că cercetarea a fost realizată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
material suplimentar
materialul suplimentar pentru acest articol poate fi găsit online la: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
date suplimentare 1 / secvența circRNA tradusă prezisă și peptida scurtă.
date suplimentare 2 | Go enrichment și Kegg enrichment results for human and Arabidopsis thaliana.
date suplimentare 3 | Compararea circrna-urilor traduse prezise cu circrna-urile traduse validate.
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: un trimmer flexibil pentru date de secvență illumina. Bioinformatică 30, 2114-2120. doi: 10.1093 / bioinformatică / btu170
PubMed Abstract / CrossRef Text Complet / Google Scholar
Brar, G. A., Weissman, J. S. (2015). Profilarea ribozomilor dezvăluie ce, când, unde și cum a sintezei proteinelor. Nat. Rev. Mol. Biol Celular. 16, 651–664. doi: 10.1038 / nrm4069
PubMed rezumat / CrossRef Text Complet / Google Scholar
Chen, L. – L., Yang, L. (2015). Reglarea biogenezei circRNA. ARN Biol. 12, 381–388. doi: 10.1080/15476286.2015.1020271
PubMed rezumat / CrossRef Text Complet / Google Scholar
Chu, Q., Zhang, X., Zhu, X., Liu, C., Mao, L., Ye, C. și colab. (2017). PlantcircBase: o bază de date pentru ARN-urile circulare ale plantelor. Mol. Planta 10, 1126-1128. doi: 10.1016 / j.molp.2017.03.003
PubMed rezumat / CrossRef text integral / Google Scholar
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jai, S. și colab. (2013). Stea: ultrarapid universal RNA-seq aligner. Bioinformatica 29, 15-21. doi: 10.1093 / Bioinformatica / bts635
PubMed rezumat / CrossRef textul integral / Google Scholar
Dong, R., Ma, X.-K., Chen, L. – L., Yang, L. (2019). „Adnotarea la nivel de genom a circrn – urilor și a alternativei lor de îmbinare/îmbinare cu conducta CIRCexplorer”, în Epitranscriptomică. Eds. Wajapeyee, N., Gupta, R. (New York, NY: Springer New York), 137-149. doi: 10.1007/978-1-4939-8808-2_10
CrossRef Text Complet / Google Scholar
Dong, R., Ma, X.-K., Li, G.-W., Yang, L. (2018). CIRCpedia v2: o bază de date actualizată pentru adnotare ARN circulară cuprinzătoare și compararea expresiilor. Genomică Proteomică Bioinf. 16, 226–233. doi: 10.1016 / j. gpb.2018.08.001
CrossRef Text Complet / Google Scholar
Gao, Y., Wang, J., Zhao, F. (2015). CIRI: un algoritm eficient și imparțial pentru identificarea ARN circulară de novo. Biol Genomului. 16, 4. doi: 10.1186 / s13059-014-0571-3
PubMed rezumat / CrossRef Text Complet / Google Scholar
Guttman, M., Russell, P., Ingolia, N. T., Weissman, J. S., Lander, E. S. (2013). Profilarea ribozomilor oferă dovezi că ARN-urile mari necodificate nu codifică proteinele. Celula 154, 240-251. doi: 10.1016 / j.celulă.2013.06.009
PubMed Rezumat / CrossRef Text Integral / Google Scholar
Hsu, P. Y., Benfey, P. N. (2018). Mic, dar puternic: peptide funcționale codificate de ORFs mici în plante. Proteomica 18, 1700038. doi: 10.1002 / pmic.201700038
CrossRef Text Complet / Google Scholar
Hsu, P. Y., Calviello, L., Wu, H.-Y. L., Li, F.-W., Rothfels, C. J., Ohler, U. și colab. (2016). Profilarea ribozomilor cu rezoluție superioară dezvăluie evenimente de traducere neanotate în Arabidopsis. Proc. Natl. Acad. Sci. 113, E7126-E7135. doi: 10.1073 / pnas.1614788113
CrossRef Text Complet / Google Scholar
Ingolia, N. T., Lareau, L. F., Weissman, J. S. (2011). Profilarea ribozomilor celulelor stem embrionare de șoarece relevă complexitatea și dinamica proteomilor de mamifere. Celula 147, 789-802. doi: 10.1016 / j.celulă.2011.10.002
PubMed rezumat / CrossRef text integral / Google Scholar
Ito, ea, Katahira, I., Vicente, F. F., da, R., Pereira, L. F. P., Lopes, F. M. (2018). BASiNET-rețeaua de secvențe biologice: un studiu de caz privind identificarea ARN-urilor de codificare și necodificare. Acizi nucleici Res. 46, e96-e96. doi: 10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093/bioinformatică / bty948
CrossRef Text Complet / Google Scholar
Kearse, M. G., Wilusz, J. E. (2017). Traducere non-AUG: un nou început pentru sinteza proteinelor în eucariote. Gene Dev. 31, 1717–1731. doi: 10.1101 / gad.305250.117
rezumat PubMed / CrossRef Text Complet / Google Scholar
Kong, L., Zhang, Y., Ye, Z.-Q., Liu, X.-Q., Zhao, S.-Q., Wei, L. și colab. (2007). CPC: evaluați potențialul de codificare a proteinelor transcrierilor folosind caracteristici de secvență și mașină vectorială de sprijin. Acizi Nucleici Res. 35, W345-W349. doi: 10.1093/nar / gkm391
rezumat PubMed / CrossRef Text Complet / Google Scholar
Langmead, B., Salzberg, S. L. (2012). Rapid gapped-citește alinierea cu papion 2. Nat. Metode 9, 357-359. doi: 10.1038 / nmeth.1923
Rezumat PubMed / CrossRef Text Complet / Google Scholar
Meng, X., Chen, Q., Zhang, P., Chen, M. (2017). CircPro: un instrument integrat pentru identificarea circrna cu potențial de codificare a proteinelor. Bioinformatica 33, 3314-3316. doi: 10.1093/Bioinformatica / btx446
PubMed rezumat / CrossRef textul integral / Google Scholar
Michel, A. M., Baranov, P. V. (2013). Profilarea ribozomilor: un monitor Hi-Def pentru sinteza proteinelor la scară largă a genomului: profilarea ribozomilor. Wiley Interdiscip. Rev. ARN 4, 473-490. doi: 10.1002 / wrna.1172
rezumat PubMed / CrossRef Text Complet / Google Scholar
Pamudurti, N. R., Bartok, O., Jens, M., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L. și colab. (2017). Traducerea CircRNAs. Mol. Celula 66, 9-21.e7. doi: 10.1016 / j. molcel.2017.02.021
Rezumat PubMed / CrossRef Text Complet / Google Scholar
Ponty, Y., Termier, M., Denise, A. (2006). GenRGenS: software pentru generarea de secvențe genomice aleatoare și structuri. Bioinformatica 22, 1534-1535. doi: 10.1093 / bioinformatică / btl113
PubMed Abstract / CrossRef Text Complet / Google Scholar
Rho, M., Tang, H., Ye, Y. (2010). FragGeneScan: prezicerea genelor în citiri scurte și predispuse la erori. Acizi nucleici Res. 38, e191-e191. doi: 10.1093/nar / gkq747
rezumat PubMed / CrossRef Text Complet / Google Scholar
A. M., S. A., Mitchell, A. J., Schwaid, A. G., Cabili, M. N., Ma, J., Levin, J. Z. și colab. (2013). Descoperirea peptidică a peptidelor codificate cu cadru de citire deschis scurt în celulele umane. Nat. Chem. Biol. 9, 59–64. doi: 10.1038 / nchembio.1120
rezumat PubMed / CrossRef Text Complet / Google Scholar
Spealman, P., Naik, A. W., mai, G. E., Kuersten, S., Freeberg, L., Murphy, R. F. și colab. (2018). Conservat non-AUG uorfs dezvăluit printr-o nouă analiză de regresie a datelor de profilare a ribozomilor. Genomul Res. 28, 214-222. doi: 10.1101 / gr.221507.117
Rezumat PubMed / CrossRef Text Complet / Google Scholar
Vicens, Q., Westhof, E. (2014). Biogeneza ARN-urilor circulare. Celula 159, 13-14. doi: 10.1016 / j.celulă.2014.09.005
PubMed rezumat / CrossRef text integral / Google Scholar
Willems, P., Ndah, E., Jonckheere, V., Stael, S., autocolant, A., Martens, L. și colab. (2017). Proteomica N-terminală a asistat la profilarea peisajului neexplorat de inițiere a traducerii în Arabidopsis thaliana. Mol. Celula. Proteomica 16, 1064-1080. doi: 10.1074 / mcp.M116. 066662
PubMed Rezumat / CrossRef Text Complet / Google Scholar
Wu, J., Mao, X., Cai, T., Luo, J., Wei, L. (2006). Kobas server: o platformă bazată pe web pentru adnotarea automată și identificarea căii. Acizi Nucleici Res. 34, W720-W724. doi: 10.1093/nar / gkl167
PubMed rezumat / CrossRef Text Complet / Google Scholar
Yang, L., Fu, J., Zhou, Y. (2018). ARN-urile circulare și rolurile lor emergente în reglarea imunității. În față. Immunol. 9, 2977. doi: 10.3389 / fimmu.2018.02977
rezumat PubMed / CrossRef Text Complet / Google Scholar
Yang, Y., Fan, X., Mao, M., cântec, X., Wu, P., Zhang, Y. și colab. (2017). Traducere extinsă a ARN-urilor circulare conduse de N6-metiladenozină. Rezoluția Celulei 27, 626-641. doi: 10.1038 / cr.2017.31
Rezumat PubMed / CrossRef Text Complet / Google Scholar
Yu, G., Wang, L.-G., Han, Y., El, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar