wprowadzenie
Dystrophies Limb-Girdle Muscular Dystrophies (LGMDs) obejmują heterogenną grupę zaburzeń charakteryzujących się postępującym zanikiem i osłabieniem mięśni bliższych kończyn (1, 2). Lgmd wykazują zmienność między-i wewnątrzrodzinną, od bardzo łagodnych postaci po ciężkie, wczesne, szybko postępujące fenotypy (2). Lgmd mogą być klasyfikowane jako autosomalnie dominujące (LGMD1) lub recesywne (LGMD2). Pierwsza grupa zwykle ma wiek dorosły i nie występuje zbyt często (<10% wszystkich Lgmd), podczas gdy te ostatnie są częstsze (1:15000) (1). Wśród postaci recesywnych LGMD2A (lub kalpainopatia) jest najczęstszym LGMD na świecie, dotyczy około 30% wszystkich przypadków Lgmd (3). Kliniczne sygnatury choroby obejmują chodzenie na palcach, wadliwy chód, trudności w bieganiu i wchodzeniu po schodach, skrzywienie szkaplerza. Często obserwuje się przykurcze stawów, skrócenie ścięgna Achillesa, skoliozę, podczas gdy mięśnie twarzy i szyi nie mają wpływu (4). Bezobjawowa Hiperkemia (5-80-krotność prawidłowych wartości CPK) u młodych pacjentów jest uważana za przedkliniczne stadium choroby i może utrzymywać się przez kilka lat (1, 5). Osłabienie mięśni występuje zwykle w wieku 15 lat, chociaż może wystąpić w wieku wcześniejszym (<12 lat) lub późniejszym (>30 lat). Postęp choroby może prowadzić do utraty ambulacji, niewydolności oddechowej i zmniejszonej pojemności życiowej płuc w zaawansowanych stadiach (6). Zaburzenia czynności serca (zaburzenia rytmu serca, zaburzenia przewodzenia serca, dysfunkcja wyrzutu lewej komory) występują tylko sporadycznie (6, 7).
rozpoznanie kalpainopatii potwierdza wykrycie mutacji patogennych w CAPN3 (15Q15 .1) (4), kodujących różne transkrypty. Jednak transkrypt pełnej długości wyraża się głównie w tkance mięśniowej (8). Kodowane białko (CAPN3) należy do nie-lizosomalnej rodziny proteaz cysteinowych zależnych od Ca++. W mięśniach CAPN3 bierze udział w” przebudowie sarcomere”, która jest niezbędna do adaptacji i wzrostu mięśni w odpowiedzi na zapotrzebowanie funkcjonalne i metaboliczne. Do tej pory opisano ponad 490 mutacji patogennych w całym CAPN3, z których większość to zmiany jednonukleotydowe (4). Mutacje w CAPN3 były związane z nieprawidłowościami mitochondrialnymi, niewydolnością wzrostu, zwiększonym stresem oksydacyjnym i dezorganizacją sarcomere, które łącznie przyczyniają się do tego, że mięsień nie jest w stanie utrzymać obciążeń, powodując tym samym degenerację włókien mięśniowych i zanik mięśni (8). Rozpoznanie kalpainopatii może być trudne ze względu na heterogeniczność genetyczną i niespecyficzność wzoru klinicznego i instrumentalnego. Rzeczywiście, rozkład osłabienia/zaniku mięśni, przerostu / pseudo-przerostu i przykurczów ścięgien są bardzo często dzielone z innymi Lgmd lub zaburzeniami nerwowo-mięśniowymi. Wzór biopsji mięśni w kalpainopatii jest na ogół nieswoisty, od łagodnych zaburzeń mięśniowych do ciężkich zmian dystroficznych. Co więcej, markery immunohistochemiczne / biochemiczne zwykle nie są wiarygodne: sygnał kalpainowy może być normalny nawet w obecności niefunkcjonalnego białka, i odwrotnie może być zmniejszony nawet w innych dystrofach mięśniowych różniących się od kalpainopatii.
dlatego zaleca się wykonanie diagnozy różnicowej w celu zapewnienia dokładnych i wiarygodnych wyników. W tym celu opracowano metody molekularnego badania genetycznego w celu potwierdzenia diagnozy kalpainopatii, w tym panel multigenowy, ekstensywny Genom (sekwencjonowanie exomu/genomu) i analiza pojedynczego genu (sekwencjonowanie bezpośrednie) (9). Wdrożenie sekwencjonowania nowej generacji (NGS) było pomocne w generowaniu danych informacyjnych, które mają być stosowane do celów diagnostycznych, predykcyjnych lub terapeutycznych (10-12). Panele genowe NGS są oparte na analizie zestawu genów związanych z konkretną chorobą lub grupą powiązanych zaburzeń, które charakteryzują się heterogenicznością genetyczną i fenotypową. Panele NGS mogą być również przydatne do wykrywania mieszanych lub złożonych fenotypów, które wynikają z dziedziczenia więcej niż jednej wady genetycznej od rodziców (13). Ogólnie rzecz biorąc, pacjenci prezentujący złożone fenotypy, którzy są negatywni wobec znanych mutacji w specjalnie zaprojektowanych diagnostycznych panelach genowych i wymagających szerokiej diagnostyki różnicowej, kwalifikują się do sekwencjonowania całego egzomu lub genomu. Sekwencjonowanie całego egzomu / genomu jest droższym podejściem w zakresie zarządzania danymi, interpretacji i kosztów analitycznych, ale zwiększa prawdopodobieństwo dostarczenia diagnozy molekularnej podejrzewanego zaburzenia genetycznego (13). W odniesieniu do Lgmd zalecane są dedykowane panele NGS, aby zapewnić wysokie wskaźniki diagnostyczne, optymalne pokrycie, czułość i specyficzność badań klinicznych (9). Panele NGS stanowią zatem jeden z najlepszych systemów ułatwiających diagnostykę różnicową, identyfikujących nowe mutacje sprawcze i wyjaśniających korelacje genotypowo-fenotypowe (14, 15).
w manuskrypcie opisano przypadek pacjenta dotkniętego LGMD2A, dla którego zastosowanie panelu NGS pozwoliło nie tylko potwierdzić diagnozę kalpainopatii (mutacje w CAPN3), ale także zidentyfikować dodatkową, nową mutację w genie LMNA związaną z kardiomiopatią rozstrzeniową. Biorąc pod uwagę te wyniki, analiza została rozszerzona na członków rodziny proband, aby zapewnić bardziej kompleksową interpretację danych analitycznych w odniesieniu do patologicznych fenotypów.
Prezentacja przypadku
charakterystyka kliniczna Probandu i jego krewnych
początek choroby w probandu został opisany w wieku 10 lat, kiedy zgłaszała obecność przerostu łydek z chodzeniem na palcach, trudnościami w bieganiu, wchodzeniu po schodach i wstawaniu. Objawy wykazały powolne, ale postępujące pogorszenie, z późniejszym zajęciem bliższej kończyny górnej. W wieku 13 lat przypadkowo odkryto wysoki poziom CPK (~8000 UI/L), Nigdy nie związany z mioglobinurią. Sukcesywnie pacjent poddawany był kilku badaniom nerwowo-mięśniowym w różnych wyspecjalizowanych ośrodkach. Wykonano dwie biopsje mięśni, pokazujące klasyczny obraz dystroficzny (włókna przerostowe/zanikowe, jądra wewnętrzne, włókna martwicze, wzrost tkanki łącznej)o niespecyficznych cechach. Zgodnie z oczekiwaniami, badania immunochemiczne i immunoblotowe były niejednoznaczne wskazując na nieprawidłowy / zredukowany sygnał α, β, γ sarkoglycan, β-dystroglikan i dystrofina tylko w włóknach martwiczych (powtarzające się immunochemia powodowały normę, również dla lamininy); immunobloty na dystrofinę i kalpain ujawniły normalne sygnały. Wiadomo jednak, że immunoblot calpain-3 nie jest całkowicie wrażliwy na diagnozę LGMD2A, ponieważ 20-30% przypadków wykazuje normalną ilość białka (1, 16-18). Ze względu na prezentację kliniczną (hiperckmia, przerost łydki/pseudo-przerost) dystrofinopatie zostały po raz pierwszy wykluczone. Ponadto analiza genów DMD (Xp21), FKRP (19q13.32) i DYSF (2P13.2) nie wykazała mutacji patogennych. Pacjent przyszedł na naszą obserwację w wieku 30 lat, wykazując zaangażowanie osiowe i obręczowe zarówno w kończynie górnej, jak i dolnej, ze znacznym wadliwym chodem i skrzydlatymi łopatkami. W kończynach dolnych osłabienie było widoczne w mięśniach czworogłowych i pośladkowych, które były znacznie zanikowe, wraz z objawami „pozornie przerostowych” (pseudo-przerostowych) mięśni łydek (ryc. 1). Nie obserwowano skurczów, mięśni i falowania. Kompletne badania pneumologiczne (spirometria i polisomnografia) i kardiologiczne (echografia, 24-godzinne monitorowanie Holtera EKG, rezonans magnetyczny serca, pełna kardiologiczna Kontrola fizyczna z ukierunkowanym wywiadem medycznym i analiza odruchów sercowo-naczyniowych) nie wykazały żadnych istotnych nieprawidłowości. Wykluczyliśmy również niedobór kwaśnej maltazy (normalne testy aktywności enzymów w limfocytach/leukocytach). Obrazowanie rezonansu magnetycznego mięśni (MRI) wykazało wyraźne zaangażowanie obręczy łopatkowej, mięśni przykręgowych, mięśni przedziału tylnego uda (ze względnym oszczędzaniem mięśni Sartoriusa i gracilisa) i nogi (w szczególności gastrocnemius medialis i soleus) (ryc. 2). Ten wzorzec zaangażowania został już opisany w literaturze dotyczącej obrazu MRI w kalpainopatii (19, 20). Ponadto badanie MRI wykazało, że mięśnie łydek były skutecznie zanikowe i charakteryzowały się naciekiem tłuszczowym, powodując „powiększenie mięśni”.”Prezentacja kliniczna, wiek wystąpienia choroby, szybkość progresji, rozkład osłabienia mięśni i wyniki badań MRI w proband były w pełni zgodne z diagnozą LGMD2A szeroko opisaną w literaturze (4, 21, 22).
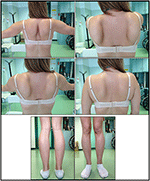
Rysunek 1. Cechy kliniczne probanda: skrzydlate łopatki i połączenie zaniku uda i pseudo-przerostu łydki.
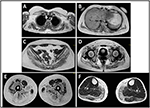
Rysunek 2. MRI mięśnia probanda: zaangażowanie obręczy kończyny górnej i mięśni przyramiennych (A, B), zaangażowanie obręczy kończyny dolnej i przedziału tylnego uda (C–E, głównie pośladków) ze względnym oszczędzaniem Sartoriusa i gracilisa oraz zaangażowanie przedziału tylnego nogi (f, głównie gastrocnemius medialis/soleus).
ocena członków rodziny proband nie ujawniła żadnej choroby nerwowo-mięśniowej w wywiadzie ani żadnych dowodów pokrewieństwa między rodzicami. Jednak matka probanda, w wieku 55 lat, przedstawiła 2 omdlenia, w których zdiagnozowano u niej bradykardię idiopatyczną (do 40 uderzeń na minutę w nocy). Rozległe badania kardiologiczne na matce probanda zdiagnozowały objawowy blok przedsionkowo-komorowy (AV) po wystąpieniu kilku epizodów omdlenia/lipotymii. Analiza wykazała obecność podstawowego bloku AV pierwszego stopnia, z kilkoma okresami ciężkiej bradykardii, głównie nocnej i często objawowej, związanej z niektórymi fazami bloku AV drugiego stopnia, zarówno typu 1, jak i 2, oraz niektórymi fazami całkowitej dysocjacji AV w nocy. Echokardiografia i badanie ergometryczne nie wykazały istotnych zmian, z wyjątkiem otworu owalnego. Matka nie przedstawiła żadnych innych objawów klinicznych, stanu predysponującego ani czynnika ryzyka. Biorąc pod uwagę jej obraz kliniczny, matka probanda została wszczepiona PMK. Ponadto historia rodzinna ujawniła, że babcia i pradziadek probandu zostały również wszczepione PMK odpowiednio w wieku 55 i 30 lat. Co więcej, brat babci probanda zmarł w wieku 51 lat na ciężką kardiomiopatię rozstrzeniową. Badania kardiologiczne przeprowadzono również na ojcu i Stryju matki probanda, które zostały całkowicie nienaruszone.
niniejsze badanie zostało zatwierdzone przez Komisję Etyki Fundacji Santa Lucia i zostało przeprowadzone zgodnie z deklaracją Helsińską. Wszyscy uczestnicy przedstawili podpisaną świadomą zgodę na analizę genetyczną, a w związku z tym wyrazili również zgodę na publikację niniejszego raportu przypadku.
badania laboratoryjne i testy diagnostyczne
genomowy DNA ekstrahowano z krwi obwodowej (400 µL) przy użyciu zestawu do ekstrakcji DNA z krwi MagPurix i systemu automatycznego ekstrakcji MagPurix (Resnova) zgodnie z instrukcjami producenta. Próbki zsekwencjonowano za pomocą ION PGM System i Ion Ampliseq Customized Panel High specific (Thermo Fisher Scientific). Wielkość panelu wynosiła 129.13 Kb, co ma być ekranowane ~99,72% całego panelu przy minimalnym pokryciu 20X. panel obejmował 18 genów, które zostały wybrane na podstawie literatury naukowej, Geneviews (www.ncbi.nlm.nih.gov/books/NBK1116/) oraz częstość występowania wariantów chorobotwórczych w populacji ogólnej. Szczegółowy opis panelu NGS przedstawiono w tabeli 1.
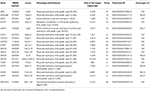
Tabela 1. Dostosowany panel NGS wykorzystywany do diagnozy LGMD.
Budowa bibliotek została wykonana przez Ion AmpliSeq™ Library Kits 2.0. Około 10 ng / µl wyjściowego DNA wykorzystano do multipleksowych reakcji PCR. Kolejno przeprowadzono dwa etapy oczyszczania (przy użyciu AMPure XP, Beckman Coulter) w celu usunięcia niepożądanych zanieczyszczeń i wykonano końcowy PCR. Etapy amplifikacji i wzbogacania wykonywały Ion PGM Hi-Q OT2 kit-400, Ion OneTouch 2 System oraz ION OneTouch ES (Thermo Fisher Scientific). Próbki były przetwarzane przez Ion PGM Hi-Q Sequencer Kit (400 bp, Thermo Fisher Scientific) i uruchamiane na Ion 316 Chip v2 (wymagane 850 przepływów) i Ion PGM Sequencer (Thermo Fisher Scientific). Wyniki analizowano przy użyciu Ion Reporter 4.6 (Thermo Fisher Scientific) i Integrated Genome Viewer (IGV). Interpretację wariantów genetycznych przeprowadzono w Human Gene Mutation Database (HGMD), Leiden Open Variation Database (LOVD), ClinVar i ExAC. Efekt funkcjonalny wykrytych wariantów oceniano za pomocą bioinformatycznych narzędzi predykcyjnych, w tym Mutation Taster, Varsome, SIFT, PolyPhen 2, SMART, Human Splicing Finder (HSF). Sekwencjonowanie bezpośrednie (BigDye Terminator v3.1, BigDyeX Terminator i ABI3130, Applied Biosystems) przeprowadzono w celu potwierdzenia wariantów genetycznych i sekwencji genomowych regionów kodujących o zasięgu <20X (Lmna, Chr1:156106052-156106076; DYSF, Chr2:71753352-71753502, Chr2:71776385-71776640; SGCB, Chr4:52904225-52904560; Sgca, chr17:48243242-48243570).
wyniki
analiza NGS proband ujawniła 3 warianty heterozygotyczne (dodatkowa rycina 1). Dwa warianty zostały zlokalizowane w capn3, mianowicie NM_000070.2 (CAPN3): c.550dela (P. Thr184Argfs) i c.1813G>C (P. Val605Leu) odpowiednio w eksonach 4 i 16. Pierwszy to delecja pojedynczego nukleotydu i jest najczęstszym (75% przypadków) wariantem chorobotwórczym w krajach europejskich (1). Zgodnie z oczekiwaniami, analiza bioinformatyczna sklasyfikowała c. 550delA (rs80338800) jako wariant utraty funkcji (p. Thr184Argfs), powodując przesunięcie ramki otwartej ramki odczytu. Narzędzia predykcyjne (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) opisywały wariant jako szkodliwy. Ponadto SMART ujawnił, że zmieniony produkt białkowy nie zawiera domeny calpain 3 i trzech motywów „EF-hands”. Pierwsza domena jest zaangażowana w szlaki sygnałowe Calpain, podczas gdy EF-hands są niezbędne do zależnej od Ca++aktywacji białka (4).
w odniesieniu do c.1813G > C, jest to nowy wariant missense (P. Val605Leu), który został przewidywany jako szkodliwy (przez mutację Taster, HSF, Varsome, PolyPhen 2, SIFT) z powodu potencjalnej zmiany splicingu. Wariant ten nie jest notowany w literaturze ani wśród internetowych baz danych i nie został znaleziony u 200 osób. Niestety, analiza za pomocą inteligentnego narzędzia nie przyniosła znaczących wyników, ponieważ wariant znajduje się w nietypowej domenie. Analiza segregacji CAPN3 wykazała, że zarówno matka, jak i wujek matki byli heterozygotyczni dla ok.550delA, podczas gdy ojciec był nosicielem c.1813g>C.
ponadto analiza NGS na probandzie wykazała obecność nowego wariantu w LMNA, mianowicie NM_170707 (LMNA): c.550C>T (S. Gln184*). Wariant ten został uznany za wariant zerowy, nie został jeszcze opisany w literaturze ani wśród internetowych baz danych i nie został znaleziony w 200 badanych. Narzędzia predykcyjne (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) opisywały patogenny wpływ tego wariantu na produkt białkowy. SMART tool poinformował, że zmienione białko LMNA nie ma domeny żarnika, która jest niezbędna do utrzymania jego struktury i funkcji. Analiza segregacji wykazała obecność c.550C> wariant t tylko u matki, co sugeruje możliwy związek z jej objawami kardiologicznymi i historią choroby w rodzinie.
zgodnie z kryteriami ustalonymi przez American College of Medical Genetics (Acmg) Standards and Guidelines (23), c.1813G> C (CAPN3) może być opisany jako prawdopodobny wariant chorobotwórczy, biorąc pod uwagę, że znajduje się w krytycznej domenie bez łagodnej zmienności (PM1); nie występuje w bazach danych ExAc, GnomAD i 1000 Genome Browser (PM2); biorąc pod uwagę recesywny model dziedziczenia, został on wykryty w trans z wariantem patogennym (PM3); wiele linii dowodów obliczeniowych potwierdza szkodliwy wpływ na gen lub produkt genowy (PP3). Jeśli chodzi o klasyfikację kliniczną c.550C>t (LMNA) przez ACMG, można go określić jako wariant patogenny, ponieważ jest wariantem zerowym powodującym utratę funkcji (p. Gln184*) w LMNA (PVS1); nie występuje w bazach danych ExAc, GnomAD i 1000 Genome Browser (PM2); znajduje się w krytycznej domenie bez łagodnej zmienności (PM1); wiele linii obliczeniowych dowodów potwierdza szkodliwy wpływ na gen lub produkt genowy (PP3).
wnioski
ten raport przypadku przedstawił pacjenta dotkniętego LGMD, który okazał się nosicielem mutacji nie tylko w CAPN3 (c.550delA i c.1813G>C), ale także w LMNA (c.550C>T). Wyniki te wyjaśniają fenotyp nerwowo-mięśniowy probanda z powodu mutacji CAPN3 i podkreślają potencjalne ryzyko zaburzeń sercowo-naczyniowych ze względu na obecność wariantu w LMNA i jej pozytywny wywiad rodzinny. Biorąc pod uwagę te wyniki, członkowie rodziny zostali poddani analizie segregacji. W szczególności matka była nosicielem odpowiednio CAPN3_c.550dela i Lmna_c.550C>T. Matkę można uznać za zdrową nosicielkę patogennej mutacji CAPN3_c.550delA związanej z kalpainopatią. Jednak obecność nowego wariantu w LMNA może tłumaczyć jej patologię sercowo-naczyniową (epizody bradykardii i omdleń). Brak objawów nerwowo-mięśniowych u matki i specyficzny obraz kliniczny probanda wykluczały możliwy związek LMNA_c.550C> T z fenotypem nerwowo-mięśniowym, szczególnie dotyczącym dystrofii mięśniowej Emery ’ ego-Dreifussa (EMD). W rzeczywistości EMD szczególnie wpływa na mięśnie vastus lateralis i biceps brachii, które są stosunkowo oszczędzone w LGM2A (24, 25).
wujek był heterozygotyczny tylko dla CAPN3_c.550delA i zgodnie z oczekiwaniami nie wykazywał żadnych problemów nerwowo-mięśniowych ani sercowo-naczyniowych. Ojciec doczekał się powieści CAPN3_c. 1813G> C i w ten sposób nie uległ zmianie. Ogólnie rzecz biorąc, analiza segregacji potwierdziła dziedziczenie trzech mutacji probanda od jej krewnych i podkreśliła znajomość kardiomiopatii, której nie można pominąć(ryc. 3).
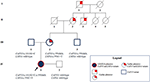
Rysunek 3. Rodowód wykazujący pozytywną znajomość fenotypu serca dziedziczonego przez rodowód matki i przekazywanie wariantów CAPN3 i LMNA przez członków rodziny.
Ogólnie Rzecz Biorąc, dane te poruszają pewne ważne kwestie. Po pierwsze, panel NGS u tego pacjenta miał kluczowe znaczenie dla osiągnięcia molekularnej diagnozy kalpainopatii, wykrywając jedną znaną mutację i drugi nowy wariant przewidywany jako patogenny. Po drugie, panel NGS umożliwił identyfikację nowego wariantu LMNA_c.550C> T w probandzie. Wynik ten był zgodny z pozytywnym wywiadem rodzinnym i danymi dotyczącymi segregacji, wyjaśniając choroby układu krążenia u matki i, co ważniejsze, zalecając bardziej szczegółową obserwację kardiologiczną w probandzie. Ważne jest, aby pamiętać, że objawy kardiologiczne wystąpiły u matki w późniejszym wieku (55 lat), więc możemy przypuszczać, że proband może nadal znajdować się w „bezobjawowej fazie kardiologicznej.”Nie można jednak wykluczyć zmiennej ekspresji fenotypowej mutacji LMNA w probandzie w porównaniu z matką. Wszystkie te dane podkreślają znaczenie zintegrowanego podejścia klinicystów i genetyków dla prawidłowej interpretacji wyników, WŁAŚCIWEGO poradnictwa genetycznego, a ostatecznie zarządzania klinicznego i obserwacji. W odniesieniu do poradnictwa genetycznego i rodzinnego, analiza NGS została również przeprowadzona na partnerze proband, w celu oszacowania ryzyka reprodukcyjnego dla pary. Partner był negatywny dla wszystkich 18 badanych genów, co oznacza, że ma 1/650 ryzyko szczątkowe, aby być zdrowym nosicielem mutacji powodujących LGMD, biorąc pod uwagę, że test jest wrażliwy na 84%. Biorąc pod uwagę profil genetyczny probanda i czułość testu NGS, ryzyko szczątkowe dla pary, aby mieć dziecko dotknięte kalpainopatią wynosi 1/1300. Z drugiej strony, warianty chorobotwórcze lmna są przenoszone według wzoru autosomalnego dominującego, co oznacza, że proband ma 50% prawdopodobieństwo posiadania dzieci heterozygotycznych. Nie można jednak z pewnością przewidzieć obrazu klinicznego potomstwa, ponieważ LMNA_c. 550C> T jest nowatorskim wariantem, a jego funkcjonalny wpływ na fenotyp jest nadal nieznany.
podsumowując, ten raport przypadku podkreśla przydatność kliniczną paneli NGS w celu zapewnienia dokładnej diagnozy LGMD2A i opisania złożonych fenotypów i chorób współistniejących pochodzących z dziedziczenia różnych mutacji w wielu genach. Jednak stosowanie NGS w praktyce klinicznej powinno być zawsze połączone z poradnią przed-i postgenetyczną, aby zapewnić jasne wyjaśnienie wyników, możliwych konsekwencji dla fenotypu pacjentów, ryzyka nawrotów w rodzinie, a także wyjaśnić możliwe nieoczekiwane wyniki.
dostępność danych
Nie wygenerowano ani nie przeanalizowano zestawów danych dla tego badania.
Deklaracja Etyczna
badanie to zostało przeprowadzone zgodnie z zaleceniami Komitetu etycznego Fundacji Santa Lucia za pisemną świadomą zgodą wszystkich uczestników. Wszystkie podmioty wyraziły pisemną świadomą zgodę zgodnie z deklaracją Helsińską. Protokół został zatwierdzony przez Komisję Etyki Fundacji Santa Lucia.
wkład autora
RC, CST, VC, GC, RG, GPA, SC, CP i JM wniosły wkład w pozyskiwanie danych, analizę i interpretację danych. CSt, RC, GC, RG, GM i EG były zaangażowane w opracowanie rękopisu. Sz, GPr, CSa i SS były zaangażowane w pozyskiwanie danych klinicznych. RC, CSt, VC, GC, RG, GPA, SC, CP, JM, SZ, GPr, GM, CSA, SS i EG wyraziły ostateczną zgodę na publikację wersji.
finansowanie
ta praca jest wspierana przez 5X 2016 Narodowe Ministerstwo Zdrowia 2.
Oświadczenie o konflikcie interesów
autorzy oświadczają, że badania zostały przeprowadzone przy braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Materiały uzupełniające
Materiały uzupełniające do tego artykułu można znaleźć w Internecie pod adresem: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. Protein and genetic diagnosis of limb girdle muscular dystrophy type 2A: the yield and the pitfalls. Nerw Mięśniowy. (2015) 52:163–73. doi: 10.1002 / mus.24682
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
2. Nigro V, Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol. (2014) 33:1–12.
PubMed Abstract / Google Scholar
3. Richard I, Hogrel JY, Stockholm D, Payan CAM, Fougerousse F, Eymard B, et al. Natural history of LGMD2A for delineating outcome measures in clinical trials. Ann Clin Transl Neurol. (2016) 3:248–65. doi: 10.1002 / acn3.287
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
4. Angelini C, Fanin M. Calpainopathy. W: Adam MP, Ardinger HH, Pagon RA editors. Generaeviews®.
PubMed Abstract / Google Scholar
5. Kyriakides T, Angelini C, Schaefer J, Sacconi S, Siciliano G, Vilchez JJ, et al. Wytyczne EFNS dotyczące diagnostycznego podejścia do pauci – lub bezobjawowej hiperckemii. Eur J Neurol. (2010) 17:767–73. doi: 10.1111 / j. 1468-1331. 2010. 03012.x
CrossRef Pełny tekst / Google Scholar
6. Mori-Yoshimura M, Segawa K, Minami N, Oya Y, Komaki H, Nonaka I, et al. Dysfunkcja krążeniowo-oddechowa u chorych z dystrofią mięśniową obręczy kończyn 2A.nerw mięśniowy. (2017) 55:465–9. doi: 10.1002 / mus.25369
CrossRef Pełny Tekst / Google Scholar
7. Okere a, Reddy SS, Gupta s, Shinnar M. kardiomiopatia u pacjenta z dystrofią mięśniową obręczy kończyn typu 2A. (2013) 6:e12–13. doi: 10.1161 / CIRCHEARTFAILURE.112.971424
CrossRef Pełny Tekst / Google Scholar
8. Kramerova I, Ermolova N, Eskin a, Hevener a, Quehenberger O, Armando AM, et al. Brak regulacji transkrypcji genów niezbędnych do adaptacji mięśni leży u podstaw dystrofii mięśniowej obręczy kończyn 2A (kalpainopatii). Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093/hmg / ddw086
PubMed Abstract / CrossRef Full Text / Google Scholar
9. Thompson R, Straub V. Limb girdle Muscular dystrophies – International collaborations for translational research. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038/nrneurol.2016.35
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
10. Strafella C, Caputo V, Galota MR, Zampatti s, Marella G, Mauriello s, et al. Zastosowanie medycyny precyzyjnej w chorobach neurodegeneracyjnych. Przedni Neurol. (2018) 9:701. doi: 10.3389/fneur.2018.00701
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
11. Cascella R, Strafella C, Caputo V, Errichiello V, Zampatti s, Milano F, et al. W kierunku zastosowania medycyny precyzyjnej w zwyrodnieniu plamki żółtej związanym z wiekiem. Prog Retin Eye Res. (2017) 63: 132-46. doi: 10.1016 / j.preteyeres.2017.11.004
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
12. Cascella r, Strafella C, Longo G, Manzo L, Ragazzo M, De Felici C, et al. Ocena ryzyka indywidualnego dla AMD z poradnictwem genetycznym, wywiadem rodzinnym i testami genetycznymi. Oko. (2017) 32:446–50. doi: 10.1038 / eye.2017.192
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
13. Adams DR, inż. Sekwencjonowanie nowej generacji do diagnozowania podejrzanych zaburzeń genetycznych. N Engl J Med. (2019) 379:1353–62. doi: 10.1056 / NEJMc1814955
PubMed Abstract / CrossRef Full Text / Google Scholar
14. Angelini C. Choroba nerwowo-mięśniowa. Diagnoza i odkrycie dystrofii mięśniowej obręczy kończyn. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038/nrneurol.2015.230
CrossRef Pełny Tekst / Google Scholar
15. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett a, Reddel SW, et al. Use of whole-exome sequencing for diagnosis of limb-girdle muscular dystrophy: outcomes and lessons learned. JAMA Neurol. (2015) 72:1424–32. doi: 10.1001 / jamaneurol.2015.2274
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
16. Milic a, Daniele N, Lochmüller H, Mora m, Comi GP, Moggio m, et al. Jedna trzecia biopsji LGMD2A ma prawidłową aktywność proteolityczną kalpain 3, co określono w teście in vitro. Neuromusc Disord. (2007) 17:148–56. doi: 10.1016 / j. nmd.2006.11.001
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
17. Lanzillo R, Aurino S, Fanin m, Aguennoz m, Vitale F, Fiorillo C, et al. Wczesna kalpainopatia z normalnym, niefunkcjonalnym poziomem kalpain 3. Dev Med Child Neurol. (2006) 48:304–6. doi: 10.1017 / S001216220600065X
PubMed Abstract / CrossRef Full Text / Google Scholar
18. Talim B, Ognibene a, Mattioli e, Richard I, Anderson LV, Merlini L. Normal calpain expression in genetically confirmed limb-girdle muscular dystrophy type 2A.Neurology. (2001) 56:692–3. doi: 10.1212 / wnl.56.5.692-a
CrossRef Pełny tekst / Google Scholar
19. Díaz-Manera J, Llauger J, Gallardo e, Illa I. MRI mięśni w dystrofii mięśniowej. Acta Myol. (2015) 34:95–108.
PubMed Abstract / Google Scholar
20. Mercuri E, Bushby K, Ricci e, Birchall DR, Pane m, Kinali m, et al. Wyniki badania MRI mięśni u pacjentów z dystrofią mięśniową obręczy kończyn z niedoborem calpain 3 (LGMD2A) i wczesnymi przykurczami. Neuromusc Disord. (2005) 15:164–71. doi: 10.1016 / j. nmd.2004.10.008
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
21. Magri F, Nigro V, Angelini C, Mongini T, Mora M, Moroni I, et al. Włoska dystrofia mięśniowa obręczy kończyn: względna częstotliwość, cechy kliniczne i diagnostyka różnicowa. Nerw Mięśniowy. (2017) 55:55–68. doi: 10.1002 / mus.25192
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
22. Chae J, Minami N, Jin y, Nakagawa m, Murayama K, Igarashi F, et al. Mutacje genu Calpain 3: genetyczne i kliniczno-patologiczne odkrycia w dystrofii mięśniowej obręczy kończyn. Neuromusc Disord. (2001) 11:547–55. doi: 10.1016 / S0960-8966(01)00197-3
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
23. Richards s, Aziz N, Bale S, Bick D, Das s, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038 / gim.2015.30
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
24. Bonne G, Mercuri E, Muchir a, Urtizberea a, Bécane HM, Recan D, et al. Kliniczne i molekularne spektrum genetyczne autosomalnej dominującej dystrofii mięśniowej Emery ’ ego-Dreifussa spowodowanej mutacjami genu lamin A/C. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170::Pomoc-ANA6 >3.0.CO; 2-J
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
25. Hong js, Ki CS, Kim JW, Suh YL, Kim js, Baek KK, et al. Zaburzenia rytmu serca, kardiomiopatia i dystrofia mięśniowa u pacjentów z dystrofią mięśniową Emery ’ ego-Dreifussa i dystrofią mięśniową obręczy kończyn typu 1B.J Korean Med Sci. (2005) 20:283–90. doi: 10.3346 / jkms.2005.20.2.283
CrossRef Full Text / Google Scholar