- wprowadzenie
- metody
- filtrowanie danych profilowania rybosomalnego
- łączenie wirtualnych genomów
- oznaczanie regionu odczytu Ribo-seq na złączu (RMRJ) cirrna
- szkolenie z modelu i klasyfikacji RMRJs
- Przewidywanie peptydów tłumaczonych przez RMRJs
- wyniki i dyskusja
- tłumaczenie cirrnas u ludzi i A. thaliana
- funkcjonalne wzbogacenie cirrnas człowieka i A. thaliana o potencjale kodowania
- Test dokładności dla CircCode
- wpływ głębokości sekwencjonowania danych Ribo-seq
- porównanie Circode z innymi narzędziami
- wnioski
- dostępność i wymagania
- Oświadczenie o dostępności danych
- author Contributions
- finansowanie
- konflikt interesów
- Materiały uzupełniające
wprowadzenie
okrągłe RNA (cirrnas) są specjalnym rodzajem niekodującej cząsteczki RNA, która stała się gorącym tematem badawczym w dziedzinie RNA i cieszy się dużym zainteresowaniem (Chen and Yang, 2015). W porównaniu z tradycyjnymi liniowymi RNA (zawierającymi końce 5′ I 3′), cząsteczki cirrna mają zwykle zamkniętą okrągłą strukturę, czyniąc je bardziej stabilnymi i mniej podatnymi na degradację (Vicens and Westhof, 2014). Chociaż istnienie cirrna jest znane od pewnego czasu, cząsteczki te były uważane za produkt uboczny splicingu RNA. Jednak wraz z rozwojem wysokoprzepustowych technologii sekwencjonowania i bioinformatyki, cirrnas stały się szeroko rozpoznawalne u zwierząt i roślin (Chen and Yang, 2015). Ostatnie badania wykazały również, że duża liczba cirrna może być przekształcona w małe peptydy w komórkach (Pamudurti et al., 2017) i mają kluczowe role pomimo ich czasami niskiego poziomu ekspresji (Hsu i Benfey, 2018; Yang et al., 2018). Pomimo coraz większej liczby cirrna są identyfikowane, ich funkcje u roślin i zwierząt na ogół pozostają do zbadania. Oprócz ich funkcji jako przynęt miRNA, cirrna mają ważny potencjał translacyjny, ale nie są dostępne żadne narzędzia do szczegółowego przewidywania zdolności translacyjnych tych cząsteczek (Jakobi i Dieterich, 2019).
istnieje kilka narzędzi do przewidywania i identyfikacji cirrna, takich jak Ciri (Gao et al., 2015), CIRCexplorer (Dong et al., 2019), CircPro (Meng et al., 2017) i circtools (Jakobi et al., 2018). Wśród nich CircPro może ujawnić przetłumaczone cirrna, obliczając potencjalny wynik tłumaczenia dla cirrna na podstawie CPC(Kong et al., 2007), który jest narzędziem do identyfikacji otwartej ramki odczytu (ORF) w danej sekwencji. Ponieważ jednak niektóre cirrna nie używają kodonu start podczas translacji (Ingolia et al., 2011; Slavoff et al., 2013; Kearse and Wilusz, 2017; Spealman et al., 2018), Zastosowanie CPC może odfiltrować niektóre prawdziwie przetłumaczone cirrna. W tym badaniu użyliśmy BASiNET (Ito et al., 2018), który jest klasyfikatorem RNA opartym na metodach uczenia maszynowego (random forest i model J48). Początkowo przekształca dane kodujące RNA (dane dodatnie) i niekodujące RNA (dane ujemne) i reprezentuje je jako złożone sieci; następnie wyodrębnia miary topologiczne tych sieci i konstruuje wektor cechowy w celu wytrenowania modelu, który jest używany do klasyfikacji zdolności kodowania cirrna. Dzięki tej metodzie unika się błędnego filtrowania przetłumaczonych cirrn, które nie są inicjowane przez AUG. Dodatkowo Technologia Ribo-seq, która opiera się na sekwencjonowaniu o wysokiej przepustowości w celu monitorowania rpfs (ribosomal protected fragments) transkryptów (Guttman et al., 2013; Brar and Weissman, 2015), można wykorzystać do określenia lokalizacji cirrnas, które są tłumaczone (Michel and Baranov, 2013). Aby zidentyfikować zdolność kodowania cirrna, opracowaliśmy narzędzie Circode, które obejmuje framework oparty na Pythonie 3 i zastosowaliśmy Circode do zbadania potencjału translacyjnego cirrna od ludzi i Arabidopsis thaliana. Nasza praca stanowi bogaty zasób do dalszego badania funkcji cirrna z możliwością kodowania.
metody
CircCode został napisany w języku programowania Python 3; używa Trimmomatic (Bolger et al., 2014), bowtie (Langmead and Salzberg, 2012) i STAR (Dobin et al., 2013), aby filtrować surowe odczyty Ribo-seq i mapować te przefiltrowane odczyty do genomu. CircCode następnie identyfikuje regiony odczytywane Ribo-seq w cirrna, które zawierają połączenia. Następnie sekwencje zmapowane kandydatami w cirrna są sortowane na podstawie klasyfikatorów (model J48) do kodowania RNA i niekodowania RNA przez BASiNET. Wreszcie, krótkie peptydy wytwarzane przez translację są identyfikowane jako potencjalne regiony kodujące cirrna. Cały proces CircCode składa się z pięciu etapów (Rysunek 1).
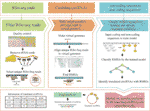
Rysunek 1 przepływ pracy CircCode. Górna warstwa reprezentuje plik wejściowy wymagany dla każdego kroku CircCode. Środkowa warstwa jest podzielona na trzy części, a każda część reprezentuje inny etap działania. Od lewej do prawej, pierwsza część reprezentuje filtrowanie danych Ribo-seq; kontrola jakości jest wykonywana przez Trimmomatic, a odczyty rRNA są usuwane przez bowtie. Druga część przedstawia kroki użyte do wytworzenia Wirtualnego genomu i dopasowania przefiltrowanych odczytów do Wirtualnego genomu za pomocą Gwiazdy. Ostatnia część przedstawia identyfikację przetłumaczonych cirrna przez uczenie maszynowe. Dolna warstwa reprezentuje ostatni etap używany do przewidywania peptydów tłumaczonych z cirrna i końcowych wyników wyjściowych, w tym informacji o przetłumaczonych cirrna i ich produktach tłumaczeniowych.
filtrowanie danych profilowania rybosomalnego
po pierwsze, niskiej jakości fragmenty i Adaptery odczytów Ribo-Seq są usuwane przez Trimmomatic z domyślnymi parametrami, aby uzyskać czyste odczyty Ribo-seq. Po drugie, te czyste odczyty Ribo-seq są mapowane do biblioteki rRNA, aby usunąć odczyty pochodzące z rRNA za pomocą bowtie. Ponieważ długości odczytu Ribo-seq są stosunkowo krótkie (zwykle mniej niż 50 bp), możliwe jest, że jeden odczyt pasuje do wielu regionów. W takim przypadku trudno jest określić, któremu regionowi odpowiada dany odczyt. Aby tego uniknąć, czyste odczyty Ribo-seq są mapowane do genomu interesującego gatunku, a odczyty, które nie są idealnie dopasowane do genomu, są uważane za ostateczne unikalne odczyty Ribo-seq.
łączenie wirtualnych genomów
Cirrna zwykle pojawiają się jako cząsteczki w kształcie pierścienia u eukariotów i można je zidentyfikować na podstawie ich tylnych połączeń. Jednak sekwencje cirrnas w pliku fasta są często w formie liniowej. W teorii, wynik wskazuje, że złącze znajduje się pomiędzy 5′ końcowym nukleotydem a 3′ końcowym nukleotydem, chociaż złącze i sekwencja w pobliżu złącza nie mogą być oglądane bezpośrednio, w ten sposób wyrównując odczyty Ribo-seq do sekwencji cirrna, w tym połączeń, w prosty sposób.
CircCode łączy sekwencję każdego cirrna w tandemie tak, że złącze dla każdego znajduje się w środku nowo skonstruowanej sekwencji. Rozdzieliliśmy również każdą jednostkę szeregową przez 100 N nukleotydów, aby uniknąć nieporozumień na etapie wyrównania sekwencji (długość każdego RPF jest mniejsza niż 50 bp). Ostatecznie otrzymaliśmy wirtualny Genom składający się tylko z kandydujących cirrna w tandemie oddzielonych 100 Ns. Ponieważ CircCode skupia się wyłącznie na dopasowaniu odczytów Ribo-seq do sekwencji cirrna, możemy zbadać potencjał kodowania cirrna poprzez mapowanie odczytów Ribo-seq do tego wirtualnego genomu, co może zaoszczędzić dużą ilość czasu obliczeniowego (wirtualny Genom jest znacznie mniejszy niż cały genom) i zwiększyć dokładność (unikając interferencji między porównaniem sekwencji cirrna w górę i w dół).
oznaczanie regionu odczytu Ribo-seq na złączu (RMRJ) cirrna
ostateczne unikalne odczyty Ribo-seq są mapowane do wcześniej utworzonego Wirtualnego genomu za pomocą Gwiazdy. Ponieważ każda tandemowa Jednostka cirrna została oddzielona przez 100 N zasad przed wytworzeniem Wirtualnego genomu, Największa długość intronu została ustawiona tak, aby nie przekraczała 10 zasad z parametrem „–alignIntronMax 10.”Ten parametr eliminuje wszelkie interakcje między różnymi cirrna w wyrównaniu sekwencji. W drugim etapie produkcji Wirtualnego genomu, CircCode przechowuje informacje o pozycyjnych połączeniach dla każdego cirrna w wirtualnym genomie. Jeśli Region odczytany z Ribo-seq w genomie wirtualnym zawiera skrzyżowanie cirrna, a liczba zmapowanych odczytów z Ribo-seq na skrzyżowaniu (NMJ) jest większa niż 3, Region odczytany z Ribo-seq na skrzyżowaniu cirrna można uznać za RMRJ, co ujawnia z grubsza przetłumaczony segment cirrna w pobliżu miejsca skrzyżowania.
szkolenie z modelu i klasyfikacji RMRJs
chociaż RMRJs mogą stanowić mocny dowód tłumaczenia, nadal istnieją pewne niedociągnięcia w tej metodzie. Ponieważ długość odczytu mapy rybosomalnej jest krótka, odczyt można porównać do niewłaściwej pozycji. Dlatego nie jest przekonujące, aby po prostu uznać region objęty czytaniem Ribo-seq za Region przetłumaczony. W tym celu wykorzystuje się metodę uczenia maszynowego do identyfikacji zdolności kodowania RMRJ. Po pierwsze, CircCode wyodrębnia kodujące RNA (dane dodatnie) i niekodujące RNA (dane ujemne) z interesującego gatunku i wykorzystuje je do treningu modelowego za pomocą różnicy w wektorach cech między kodowaniem A niekodującymi RNA. CircCode wykorzystuje następnie wytrenowany model do klasyfikacji RMRJs uzyskanych w poprzednim kroku przez BASiNET. Jeśli rmrj cirrna jest rozpoznawany jako kodujący RNA, to ten cirrna może być zidentyfikowany jako przetłumaczony cirrna.
Przewidywanie peptydów tłumaczonych przez RMRJs
ponieważ ekspresja cirrna w organizmach jest niska, dane Ribo-seq nie pokazują dokładnej 3-nt okresowości wyraźnie w przypadku mniejszej liczby RPF. Dlatego trudno jest określić dokładne miejsce rozpoczęcia tłumaczenia przetłumaczonej cirrna. Ze względu na obecność kodonu stop w niektórych Rmrj i ponieważ kodon start jest trudny do określenia, metoda znalezienia ORF na podstawie kodonu start i kodonu stop nie jest wykonalna.
aby określić prawdziwe regiony tłumaczenia tych cirrna i wygenerować końcowy produkt tłumaczenia, FragGeneScan (Rho et al., 2010), który może przewidywać regiony kodujące białka w fragmentowanych genach i genach z przesunięciami ramek, jest używany do oznaczania translowanych peptydów wytwarzanych przez cirrna.
aby uniknąć uciążliwego uruchamiania procesu, wszystkie modele mogą być wywoływane przez skrypt powłoki; użytkownik może po prostu wypełnić podany plik konfiguracyjny i wprowadzić go do skryptu, a cały proces przewidywania przetłumaczonych cirrnas zostanie uruchomiony. Ponadto CircCode może być uruchamiany oddzielnie, krok po kroku, tak aby użytkownik mógł dostosować parametry w środku procedury i wyświetlić wyniki każdego kroku zgodnie z potrzebami.
wyniki i dyskusja
po przetestowaniu na wielu komputerach okazało się, że CircCode działa pomyślnie z zainstalowanymi wymaganymi zależnościami. Aby przetestować wydajność CircCode, użyliśmy danych dla ludzi i A. thaliana do przewidywania cirrna z potencjałem translacyjnym. Wyniki porównano z cirrna, które zostały zweryfikowane doświadczalnie jako potwierdzenie. Następnie przetestowaliśmy wartość false discovery rate (FDR) CircCode. Użyliśmy GenRGenS (Ponty et al., 2006) w celu wygenerowania zestawu danych do testów w oparciu o znane przetłumaczone cirrna i potwierdziło, że wartość FDR mieści się w akceptowalnym zakresie i na niskim poziomie. Na koniec oceniliśmy wpływ różnych głębokości sekwencjonowania danych Ribo-seq na przewidywania CircCode i porównaliśmy CircCode z innym oprogramowaniem.
tłumaczenie cirrnas u ludzi i A. thaliana
aby zastosować narzędzie CircCode do rzeczywistych danych, najpierw pobraliśmy pliki, w tym ludzki genom referencyjny GRCh38, adnotację genomu i ludzki rRNA, z Ensembl. W przypadku A. thaliana genomy referencyjne (TAIR10), pliki adnotacji genomu i odpowiadające im sekwencje rRNA zostały pobrane z roślin Ensembl. Dane Ribo-seq dla ludzi i A. thaliana zostały pobrane z RPFdb (numery akcesyjne: GSE96643, GSE81295, GSE88794) (Hsu et al., 2016; Willems et al., 2017), a wszystkie kandydujące cirrnas z human i A. thaliana zostały pobrane z CIRCPedia v2 (Dong et al., 2018) i PlantcircBase, odpowiednio (Chu et al., 2017). Ostatecznie zidentyfikowaliśmy 3610 przetłumaczonych cirrnas od człowieka i 1569 przetłumaczonych cirrnas od A. thaliana za pomocą CircCode (dane uzupełniające 1).
funkcjonalne wzbogacenie cirrnas człowieka i A. thaliana o potencjale kodowania
wykorzystując wyniki Circode dla człowieka i A. thaliana, narzędzie online KOBAS 3.0 (Wu et al., 2006) został zatrudniony do przypisania tych przetłumaczonych cirrnas na podstawie ich genów macierzystych. Ponadto przeprowadziliśmy analizę funkcjonalną GO (Gene Ontology) i analizę wzbogacenia KEGG (Kyoto Encyclopedia of Genes and Genomes) dla tych przetłumaczonych cirrna przy użyciu klasterprofiler pakietu R (YU et al., 2012).
wyniki KEGG wykazały, że ludzkie cirrna zostały wzbogacone w przetwarzanie białka w szlaku retikulum endoplazmatycznego, szlaku metabolizmu węgla i Szlaku transportu RNA. Analiza GO wykazała udział ludzkich translowanych cirrna w regulacji wiązania cząsteczek, aktywności ATPazy i innych procesów biologicznych związanych ze splicingiem RNA. Ponadto przetłumaczone cirrna A. thaliana są wzbogacone w szlaki związane z odpornością na stres, co sugeruje, że odgrywają istotną rolę w tym procesie (dane uzupełniające 2).
Test dokładności dla CircCode
aby zbadać dokładność CircCode, zastosowano sekwencje testowe generowane przez GenRGenS, które wykorzystują Ukryty model Markowa do wytworzenia sekwencji o tych samych właściwościach sekwencji (takich jak częstotliwości różnych nukleotydów, różnych kodonów i różnych nukleotydów na początku sekwencji).
do tego badania wykorzystaliśmy wcześniej opublikowane ludzkie przetłumaczone cirrna (Yang et al., 2017) jako Wejście dla GenRGenS i wygenerował 10 000 sekwencji do testowania Circode. Powtórzyliśmy test 10 razy, a średnio za każdym razem przewidywano 27 przetłumaczonych cirrna. Wartość FDR została obliczona na 0,0027, czyli znacznie mniej niż 0,05, co wskazuje, że przewidywane wyniki są wiarygodne.
ponadto porównaliśmy przetłumaczone cirrna od ludzi zidentyfikowanych przez Circode ze zweryfikowanymi danymi cirrna związanymi z polisomem (Yang et al., 2017). Wśród nich 60% cirrna zostało zidentyfikowanych przez CircCode (dane uzupełniające 3).
wpływ głębokości sekwencjonowania danych Ribo-seq
aby zbadać wpływ głębokości sekwencjonowania danych Ribo-seq na wyniki identyfikacji kodu Cyrkodowego, najpierw przetestowaliśmy wpływ głębokości sekwencjonowania na liczbę przetłumaczonych cirrna (Fig.2A). Gdy głębokość sekwencjonowania była niska, przewidywana liczba przetłumaczonych cirrna była niska, a liczba przetłumaczonych cirrna wzrastała wraz ze wzrostem głębokości sekwencjonowania. Liczba transponowanych cirrna stała się stabilna, gdy głębokość sekwencjonowania osiągnęła nie mniej niż 10× liniowe pokrycie transkryptu.

fig.2 (A) wpływ głębokości sekwencjonowania danych Ribo-seq na przewidywaną liczbę przetransmitowanych cirrna. B) wpływ numeru odczytu złącza (JRN) na czułość kodu cyrkulacyjnego na różnych głębokościach sekwencjonowania.
po drugie oceniono również wpływ NMJ na czułość na różnych głębokościach sekwencjonowania (Fig.2b). Wyniki wykazały, że NMJ miał mniejszy wpływ na czułość wraz ze wzrostem głębokości sekwencjonowania. CircCode miał również wyższą czułość przy użyciu danych Ribo-seq z większą głębią sekwencjonowania.
porównanie Circode z innymi narzędziami
aby porównać Circode z innymi narzędziami, takimi jak CircPro, ten sam zestaw danych Ribo-seq (SRR3495999) A. thaliana został użyty do identyfikacji przetłumaczonych circrna przy użyciu sześciu procesorów z 16 gigabajtami pamięci RAM. CircPro zidentyfikował 44 przetłumaczone cirrna w 13 min, podczas gdy CircCode zidentyfikował 76 przetłumaczone cirrna w 20 min. Tak więc, CircCode jest bardziej czuły niż CircPro na tym samym poziomie sprzętu komputerowego, ale zajmuje więcej czasu. CircPro jest zwięzły i mniej czasochłonny niż CircCode, ale Circode może zidentyfikować więcej cirrna z możliwością kodowania niż CircPro.
wnioski
Cirrnas odgrywają ważną rolę w biologii i kluczowe jest dokładne zidentyfikowanie cirrnas ze zdolnością kodowania do dalszych badań. Opierając się na Pythonie 3, opracowaliśmy Circode, łatwe w użyciu narzędzie wiersza poleceń, które ma wysoką czułość do identyfikacji przetłumaczonych cirrna z odczytów Ribo-Seq z dużą dokładnością. CircCode wykazuje dobrą wydajność zarówno u roślin, jak i zwierząt. Przyszłe prace dodadzą do CircCode analizę kolejnych znaków poprzez wizualizację każdego kroku procesu i optymalizację dokładności prognozy.
dostępność i wymagania
CircCode jest dostępny pod adresem https://github.com/PSSUN/CircCode; system operacyjny(y): Linux, języki programowania: Python 3 i R; inne wymagania: bedtools (wersja 2.20.0 lub nowsza), bowtie, STAR, Pakiety Python 3 (Biopython, Pandas, rpy2), R-packages (BASiNET, Biostrings). Pakiety instalacyjne dla wszystkich wymaganych programów są dostępne na stronie głównej CircCode. Użytkownicy nie muszą pobierać ich indywidualnie. Strona główna CircCode zawiera również szczegółowe instrukcje obsługi w celach informacyjnych. Narzędzie jest dostępne bezpłatnie. Nie ma ograniczeń w stosowaniu przez nieakademików.
Oświadczenie o dostępności danych
wszystkie istotne dane znajdują się w rękopisie i jego pomocniczych plikach informacyjnych.
Conceptualization: PS, GL. Dobór danych: PS, GL. Analiza formalna: PS, GL. Writing – Original Draft: PS, GL. Pisanie-recenzja i montaż: PS, GL.
finansowanie
praca ta była wspierana przez granty z National Natural Science Foundation Of China (grant nr 31770333, 31370329 i 11631012), Program for New Century Excellent Talents in University (NCET-12-0896) oraz fundusze na badania podstawowe dla Uniwersytetów centralnych (nr 31770333, 31370329 i 11631012). GK201403004). Agencje finansujące nie miały żadnej roli w badaniu, jego projektowaniu, zbieraniu i analizie danych, decyzji o publikacji lub przygotowaniu manuskryptu. Fundatorzy nie mieli żadnej roli w projektowaniu badań, zbieraniu i analizie danych, decyzji o publikacji lub przygotowaniu rękopisu.
konflikt interesów
autorzy oświadczają, że badania zostały przeprowadzone przy braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Materiały uzupełniające
Materiały uzupełniające do tego artykułu można znaleźć w Internecie pod adresem: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
dane uzupełniające 1 / Sekwencja przewidywanego przetłumaczonego cirrna i krótkiego peptydu.
dane uzupełniające 2 / Go wzbogacanie i wyniki wzbogacania KEGG dla ludzi i Arabidopsis thaliana.
dane uzupełniające
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: elastyczny trymer do danych sekwencji illumina. Bioinformatyka 30, 2114-2120. doi: 10.1093/bioinformatics / btu170
PubMed Abstract / CrossRef Full Text / Google Scholar
Brar, G. A., Weissman, J. S. (2015). Profilowanie rybosomów ujawnia, co, kiedy, gdzie i jak syntezy białek. Nat. Ks. Mol. Cell Biol. 16, 651–664. doi: 10.1038 / nrm4069
PubMed Abstract / CrossRef Full Text / Google Scholar
Chen, L.-L., Yang, L. (2015). Regulacja biogenezy cirrna. RNA Biol. 12, 381–388. doi: 10.1080/15476286.2015.1020271
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
Chu, Q., Zhang, X., Zhu, X., Liu, C., Mao, L., Ye, C., et al. (2017). PlantcircBase: baza danych dla okrągłych RNA roślin. Mol. Roślina 10, 1126-1128. doi: 10.1016 / j.molp.2017.03.003
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). Gwiazda: ultraszybki uniwersalny RNA-Seq aligner. Bioinformatyka 29, 15-21. doi: 10.1093/bioinformatics / bts635
PubMed Abstract / CrossRef Full Text / Google Scholar
Dong, R., Ma, X.-K., Chen, L.-L., Yang, L. (2019). „Genome-wide annotation of cirrna and their alternative back-splicing / splicing with CIRCexplorer Pipeline,” in Epitranscriptomics. Eds. Wajapeyee, N., Gupta, R. (New York, NY: Springer New York), 137-149. doi: 10.1007/978-1-4939-8808-2_10
CrossRef Pełny tekst / Google Scholar
Dong, R., Ma, X.-K., Li, G.-W., Yang, L. (2018). CIRCpedia v2: Zaktualizowana baza danych do kompleksowej okrągłej adnotacji RNA i porównywania ekspresji. Genomika Proteomika Bioinf. 16, 226–233. doi: 10.1016 / j.gpb.2018.08.001
CrossRef Pełny Tekst / Google Scholar
Gao, Y., Wang, J., Zhao, F. (2015). CIRI: wydajny i bezstronny algorytm do De novo circular RNA identification. Genom Biol. 16, 4. doi: 10.1186 / s13059-014-0571-3
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
Guttman, M., Russell, P., Ingolia, N. T., Weissman, J. S., Lander, E. S. (2013). Profilowanie rybosomów dostarcza dowodów na to, że duże niekodujące RNA nie kodują białek. Cela 154, 240-251. doi: 10.1016 / j.cell.2013.06.009
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Hsu, P. Y., Benfey, P. N. (2018). Mały, ale potężny: funkcjonalne peptydy kodowane przez małe ORF w roślinach. / Align = „left” / 1700038 doi: 10.1002 / pmic.201700038
CrossRef Pełny tekst / Google Scholar
Hsu, P. Y., Calviello, L., Wu,H.-Y. L., Li, F.-W., Rothfels, C. J., Ohler, U., et al. (2016). Nadrozdzielcze profilowanie rybosomów ujawnia niezidentyfikowane wydarzenia translacyjne u Arabidopsis. Proc. Natl. Acad. Sci. 113, E7126-E7135. doi: 10.1073 / pnas.1614788113
CrossRef Pełny Tekst / Google Scholar
Ingolia, N. T., Lareau, L. F., Weissman, J. S. (2011). Profilowanie rybosomów zarodkowych komórek macierzystych myszy ujawnia złożoność i dynamikę proteomów ssaków. Kom. 147, 789-802. doi: 10.1016 / j.cell.2011.10.002
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
ito, E. A., Katahira, I., Vicente, F. F., da, R., Pereira, L. F. P., Lopes, F. M. (2018). BASiNET – Biological Sequences NETwork: a case study on coding and non-coding RNA identification. Kwasy nukleinowe Res.46, e96–e96. doi: 10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093/bioinformatyka / bty948
CrossRef Pełny tekst / Google Scholar
Kearse, M. G., Wilusz, J. E. (2017). Tłumaczenie Non-AUG: nowy początek syntezy białek u eukariotów. Genes Dev. 31, 1717–1731. doi: 10.1101 / gad.305250.117
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Kong, L., Zhang, Y., Ye,Z.-Q., Liu, X.-Q., Zhao, S.-Q., Wei, L., et al. (2007). CPC: ocena potencjału kodowania białek transkryptów przy użyciu funkcji sekwencji i maszyny wektorów pomocniczych. Kwasy Nukleinowe Res. 35, W345–W349. doi: 10.1093/nar / gkm391
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Langmead, B., Salzberg, S. L. (2012). Fast gapped-Czytaj wyrównanie z Bowtie 2. Nat. Metody 9, 357-359. doi: 10.1038 / nmeth.1923
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Meng, X., Chen, Q., Zhang, P., Chen, M. (2017). CircPro: zintegrowane narzędzie do identyfikacji cirrna z potencjałem kodowania białka. Bioinformatyka 33, 3314-3316. doi: 10.1093/bioinformatyka / btx446
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Michał, A. M., Baranov, P. V. (2013). Profilowanie rybosomów: monitor Hi-Def do syntezy białek w skali całego genomu: profilowanie rybosomów. Wiley Interdiscip. Rev. RNA 4, 473-490. doi: 10.1002 / wrna.1172
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Pamudurti, N. R., Bartok, O., Jens, M., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., et al. (2017). Tłumaczenie Cirrnas. Mol. Cela 66, 9-21.e7. doi: 10.1016 / j.molcel.2017.02.021
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Ponty, Y., Termier, M., Denise, A. (2006). GenRGenS: oprogramowanie do generowania losowych sekwencji i struktur genomowych. Bioinformatyka 22, 1534-1535. doi: 10.1093/bioinformatics / btl113
PubMed Abstract / CrossRef Full Text / Google Scholar
Rho, M., Tang, H., Ye, Y. (2010). FragGeneScan: przewidywanie genów w krótkich i podatnych na błędy czytaniach. Kwasy nukleinowe Res. 38, e191-e191. doi: 10.1093/nar / gkq747
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
2010-02-12 11: 45: 45 (2013). Peptydomiczne odkrycie krótkich otwartych ramek odczytu kodowanych peptydów w ludzkich komórkach. Nat. Chem. Biol. 9, 59–64. doi: 10.1038 / nchembio.1120
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Spealman, P., Naik, A. W., May, G. E., Kuersten, S., Freeberg, L., Murphy, R. F., et al. (2018). Zachowane uorfy nie-AUG ujawnione przez nowatorską analizę regresji danych profilowania rybosomów. Genome Res. 28, 214-222. doi: 10.1101 / gr.221507.117
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
Vicens, Q., Westhof, E. (2014). Biogeneza kolistych RNA. Cela 159, 13-14. doi: 10.1016 / j.cell.2014.09.005
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Willems, P., Ndah, E., Jonckheere, V., Stael, S., Sticker, A., Martens, L., et al. (2017). Proteomika N-końcowa wspomagała profilowanie niezbadanego krajobrazu inicjacji translacji u Arabidopsis thaliana. Mol. Cell. Proteomics 16, 1064-1080. 10.1074 / mcp.M116. 066662
PubMed Streszczenie | CrossRef Pełny Tekst / Google Scholar
Wu, J., Mao, X., Cai, T., Luo, J., Wei, L. (2006). KOBAS server: internetowa platforma do automatycznej adnotacji i identyfikacji ścieżek. Kwasy Nukleinowe Res. 34, W720–W724. doi: 10.1093/nar / gkl167
PubMed Abstract / CrossRef Full Text / Google Scholar
Yang, L., Fu, J., Zhou, Y. (2018). Okrągłe RNA i ich pojawiające się role w regulacji immunologicznej. Przód. Immunol. 9, 2977. doi: 10.3389 / fimmu.2018.02977
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
Yang, Y., Fan, X., Mao, M., Song, X., Wu, P., Zhang, Y., et al. (2017). Obszerna translacja okrągłych RNA napędzanych przez N6-metyloadenozynę. Kom. 27, 626-641. doi: 10.1038 / cr.2017.31
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
YU, G., Wang, L.-G., Han, Y., He, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar