med sår av sunt vev utfolder en forutsigbar progresjon av fysiologiske hendelser. Denne progresjonen kan deles inn i faser av betennelse, proliferasjon og modning. Hver fase er preget av sekvensiell utarbeidelse av karakteristiske cytokiner av bestemte celler. Se bildene nedenfor.
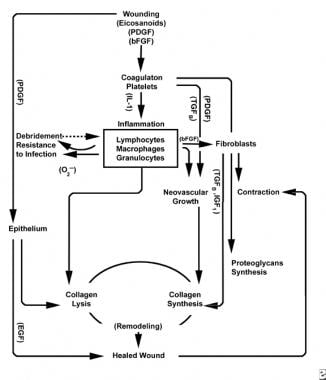 Ordninger av sårhelingsprosessen.
Ordninger av sårhelingsprosessen. 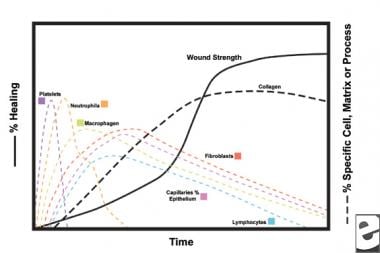 Cellulære egenskaper av sårhelingsprosessen.
Cellulære egenskaper av sårhelingsprosessen. den inflammatoriske fasen
den inflammatoriske fasen lanserer samtidig hemostatiske mekanismer og veier som skaper klinisk gjenkjennelige kardinale tegn på betennelse: rubor (rødhet), calor (varme), svulst (hevelse), dolor (smerte) og functio laesa (tap av funksjon).
Skade på vaskulært vev initierer den ytre koagulasjonskaskaden ved å frigjøre intracellulær kalsium og vevsfaktor som aktiverer faktor VII. den resulterende fibrinpluggen oppnår hemostase hjulpet av refleks vasokonstriksjon. Denne pluggen fungerer som et gitter for aggregering av blodplater, den vanligste og» signatur » celletypen av den tidlige inflammatoriske fasen.
Blodplater utarbeider en rekke proinflammatoriske stoffer, som adenosindifosfat, vevsvekstfaktor beta (TGF-ß) og blodplatederiverte vekstfaktorer (PDGF). Disse vekstfaktorene virker på omgivende celler og stimulerer kjemotaksis av nøytrofiler, monocytter og fibroblaster til skadeområdet.
Skadet vev, gjennom aktivert fosfolipase A, katalyserer samtidig arakidonsyrer for å produsere vasoaktive prostaglandiner og tromboxan, kollektivt kjent som eikosanoider. Eikosanoider medierer aktivitet som påvirker blodplatepluggdannelse, vaskulær permeabilitet og cellulær kjemotaksis for å påvirke sårheling. For eksempel medierer tromboxan A2 vasokonstriksjon og blodplateaggregering.
etter innledende vasokonstriksjon manifesterer de klassiske tegn på betennelse fra økt vaskulær permeabilitet. Rubor er et resultat av vasodilatasjon, mediert av prostacyklin (PGI2), prostaglandin A (PGA), prostaglandin D (PGD) og prostaglandin E (PGE). Tumor og calor utvikles som vaskulære endoteliale hull forstørres, slik at utgangen av plasmaprotein og væske inn i interstitialrommet. Disse endringene forsterkes AV pge2 Og prostaglandin F2a (PGF2a) og tillater inntrengning av inflammatoriske celler i skadeområdet, inkludert celler som utdyper. Dolor er merket SOM PGI2, PGE, OG PGE2 handle på perifere nociceptorer.
i den andre fasen av den inflammatoriske fasen erstatter leukocytter blodplater som den dominerende celletypen, tiltrukket av kjemotaksis. Hvite blodlegemer (Wbc) er de dominerende cellene for de første 3 dagene etter såret; deres tall topp på ca 48 timer. Polymorfonukleocytter (pmn) er de første som begynner bakteriedrepende aktiviteter ved bruk av inflammatoriske mediatorer og oksygenfrie radikale metabolitter. Imidlertid kan normal sårheling oppstå uten PMNs. En annen leukocyt, hjelper T-cellen, utdyper interleukin – 2 (IL–2). IL-2 fremmer ytterligere t-celleproliferasjon for å øke den immunogene responsen på skade.
da pmn leukocytter begynner å avta etter 24-36 timer, kommer sirkulerende monocytter inn i såret og modnes i vevsmakrofager. Disse cellene debride såret på mikroskopisk nivå og produsere en rekke viktige stoffer, SLIK SOM IL-1 og basisk fibroblast vekstfaktor (bFGF). IL-1 stimulerer proliferasjonen av inflammatoriske celler og fremmer angiogenese gjennom replikasjon av endotelceller. bFGF er en kjemotaktisk og mitogen faktor for fibroblaster og endotelceller. I motsetning Til Pmn, makrofag uttømming alvorlig svekker sårtilheling, som debridement, fibroblast proliferasjon, og angiogenese alle avta.
Mot slutten av den inflammatoriske syklusen interagerer det utviklende miljøet av eikosanoider i såret med celletypene som er tilstede, noe som resulterer i fibroblastsyntese av kollagen og grunnstoff (fra økt forhold Mellom PGF2a OG PGE2). I tillegg er de makrofagavledede vekstfaktorene nå på optimale nivåer, og påvirker sterkt tilstrømningen av fibroblaster og deretter keratinocytter og endotelceller inn i såret. Som mononukleære celler fortsetter å erstatte Wbc og makrofager, begynner den proliferative fasen.
den proliferative fasen
to til tre dager etter såret migrerer fibroblaster innover fra sårmarginer over den fibrinøse matrisen som er etablert under den inflammatoriske fasen. I løpet av den første uken begynner fibroblaster å produsere glykosaminoglykaner og proteoglykaner, grunnstoffet for granulasjonsvev, samt kollagen, som respons på makrofag-syntetisert bFGF og TGF-ß, SAMT PDGF.
Fibroblaster blir snart den dominerende celletypen, toppet på 1-2 uker. De genererer ikke bare kollagenmolekyler, men også cytokiner som PDGF, TGF-ß, bFGF, keratinocyttvekstfaktor og insulinlignende vekstfaktor-1. Fibroblaster samler også kollagenmolekyler i fibre, som er tverrbundet og organisert i bunter. Kollagen er hovedkomponenten i akutt sårbindevev, med nettoproduksjon fortsetter de neste 6 ukene. Det økende innholdet av sårkollagen korrelerer med økende strekkstyrke.
Keratinocytter og endotelceller sprer seg også i løpet av denne tiden, og produserer til slutt autokrine vekstfaktorer som opprettholder veksten. Endotelial ekspansjon bidrar til angiogenese, da intakte kar genererer knopper i granulasjonsvev. Neovaskularisering letter veksten av den fremrykkende linjen av fibroblaster inn i såret, og gir dem nødvendige næringsstoffer og cytokiner.
Nedbrytning av fibrinklumpen og foreløpig matrise ledsages av avsetning av granulasjonsvev (grunnstoff, kollagen, kapillærer), som fortsetter til såret er dekket. Reduksjon av hyaluronsyre (i grunnstoff) nivåer og økende kondroitinsulfatnivåer reduserer fibroblastmigrasjon og proliferasjon mens det induserer fibroblastdifferensiering, overgang til modningsfasen av sårheling.
modningsfasen
i de første 6 ukene dominerer ny kollagenproduksjon sårhelingsprosessen, avsatt tilfeldig i akutt sårgranulasjonsvev. Når såret modnes, blir kollagen omformet til en mer organisert struktur med økt strekkstyrke. Gradvis erstatter type i kollagen TYPE III til det normale hudforholdet på 4: 1 oppnås. Som remodeling fortsetter, oppnår matrix metalloproteinase kollagenolyse en steady state med kollagen syntese. Strekkfasthet platåer på 80% av den opprinnelige styrken ca 1 år etterskade.
Overfladisk til denne aktiviteten fortsetter epitelceller å migrere innover fra sårkanten til defekten er dekket. På dette punktet induserer kontaktinhibering transformasjon av fibroblaster til myofibroblaster, som inneholder kontraktile aktinfibre. Sårkontraksjon følger, erstatter skadet vevsvolum med nytt vev, selv om myofibroblastens nøyaktige rolle ikke er fullstendig klarlagt.
Sårheling
Akutte sår går vanligvis gjennom en ordnet og rettidig reparativ prosess som resulterer i en varig restaurering av anatomisk og funksjonell integritet. Imidlertid kan forskjellige fysiologiske og mekaniske faktorer svekke helbredelsesresponsen, noe som resulterer i et kronisk sår som ikke klarer å fortsette gjennom den vanlige trinnvise progresjonen. Lokal infeksjon, hypoksi, traumer, fremmedlegemer eller systemiske problemer som diabetes mellitus, underernæring, immundefekt eller medisiner er oftest ansvarlige.
alle sår er forurenset, men mest vellykket motstå invasiv infeksjon. Når konsentrasjonen overstiger 100.000 (105) organismer per gram vev eller immunsystemet blir kompromittert, oppstår infeksjon ofte. Cellulitt forlenger den inflammatoriske fasen ved å opprettholde høye nivåer av proinflammatoriske cytokiner og vevsproteaser, som nedbryter granulasjonsvev og vevsvekstfaktorer, og ved å forsinke kollagenavsetning.
Debridement (kirurgisk, enzymatisk og/eller ved bandasjeskift) og antibiotika er bærebjelkene i antibiotikabehandling. Debridement fjerner devitalized vev, som kan være en kilde til endotoksiner som hemmer fibroblast og keratinocyttmigrasjon i såret. Fremmedlegemer kan også kreve fjerning, da tilstedeværelsen av en silke sutur reduserer antall bakterier som kreves for å anspore infeksjon 10.000 ganger. (For en detaljert beskrivelse av teknikk, se Medscape Referanseartikkel Sår Fremmedlegeme Fjerning.)
Cellulær hypoksi forsinker sårheling på ulike måter. Kollagenfibril tverrbinding krever oksygen til hydroksylatprolin og lysin og svikter når vevstrykket er under 40 mm Hg. Den bakteriedrepende styrken av leukocyttoksidativ fosforylering lider også i et hypoksisk miljø, og reduserer terskelen for infeksjon. Tiltak for å forbedre oksygentilførselen avhenger av etiologien. Tobaksbruk, som forårsaker vasokonstriksjon og øker blodplateadherence, bør stoppes. Angioplastikk eller arteriell bypass-podning kan være nødvendig for perifer vaskulær sykdom. Tilleggsforanstaltninger for å forbedre systemisk perfusjon i tilfeller av hjertesvikt kan være indisert. Hematokrit verdi mindre enn 15% bør behandles og euvolemia gjenopprettes, etter behov. Venøs stasis eller lymfatisk insuffisiens kan forbedres med trykkplagg.
Systemisk sykdom kan dramatisk forlenge eller avbryte sårheling. Glykosylering i diabetes mellitus svekker nøytrofil-og makrofagfagocytose av bakterier, forlenger den inflammatoriske fasen. Den proliferative fasen er også langvarig i samme sykdom som erytrocytter blir mindre bøyelige og mindre i stand til å levere oksygen til såret for vevmetabolisme og kollagensyntese.
Underernæring resulterer i redusert fibroblastproliferasjon, svekket neovaskularisering og redusert cellulær og humoral immunitet. Sår utøver økte metabolske krav, spesielt i granulasjonsvev. Aminosyrer som metionin, prolin, glycin og lysin er essensielle for normal cellefunksjon og reparasjon av kutane sår. Fettsyrer er kritiske bestanddeler av cellemembraner og er substratet for eikosanoider som formidler den inflammatoriske prosessen. Essensielle fettsyrer linolensyre og linolsyre må leveres i dietten, da menneskekroppen ikke er i stand til de novo syntese av disse molekylene.
Tilstrekkelige vitaminer og mineraler må være tilgjengelige for cellemetabolisme, som fungerer som cellulære signaler og kofaktorer. Vitamin C (askorbinsyre) og jern er nødvendig for hydroksylering av lysin og prolin, som krysser og stabiliserer trippelhelixstrukturen av kollagen; kobber spiller også en rolle i å stabilisere kollagen. Vitamin a (retinsyre) spiller en viktig rolle i å modulere kollagenproduksjon og nedbrytning og er spesielt viktig i epithelialisering. En potent antioksidant, vitamin e (alfa-tokoferol) ser ut til å akselerere dermal og beinheling hos dyr, og tilskudd kan ha en rolle hos mennesker. Spormetall, spesielt sink, mangel er også forbundet med dårlig sårheling; dette bør etterfylles, etter behov.
Ovid skrev angivelig, » medisiner noen ganger helbrede, noen ganger drepe.»Dette er sikkert sant om sårheling. Kortikosteroider slår prosessene i hele inflammatorisk fase. Vitamin a (lokalt eller 25 000 IE/d oralt) reduserer de skadelige helbredende effektene av kortikosteroider, men hepatotoksisitet kan skyldes langvarig bruk (dvs. >1 mo). Ikke-steroide antiinflammatoriske medisiner (NSAIDs) forstyrrer også arakidonsyremetabolismen og derfor sårheling. I Tillegg Hemmer NSAIDs blodplatefunksjonen, en av de tidligste prosessene i den inflammatoriske fasen.
En studie Av Sutcliffe et al antydet at oppregulering av gapet krysset protein connexin er vanlig for kroniske sår. Undersøke connexin i tre typer sår-venøse ben, diabetessår, og trykksår-etterforskerne fant at hver type sår vises oppregulering av epidermal connexin 43, connexin 26, og connexin 30, samt dermal connexin 43.