Innledning
Lgmds Inkluderer en heterogen gruppe lidelser preget av progressiv sløsing og svakhet i de proksimale lemmer-girdles musklene (1, 2). LGMDs viser inter-og intrafamilial variabilitet, alt fra svært milde former, til alvorlige, tidlig begynnende, raskt progressive fenotyper (2). Lgmd kan klassifiseres som autosomal dominant (lgmd1) eller recessiv (LGMD2). Den første gruppen har vanligvis en voksen alder av debut og er ikke veldig vanlig (<10% Av Alle LGMDs), mens sistnevnte er hyppigere (1:15000) (1). Blant recessive former ER LGMD2A (eller calpainopathy) den vanligste lgmd over hele verden, noe som påvirker omtrent 30% Av Alle lgmds-tilfeller (3). Kliniske signaturer av sykdom inkluderer tiptoe walking, waddling gang, vanskeligheter med å løpe og klatre trapper, scapular winging. Felles kontrakturer, Akillesseneforkortelse, skoliose observeres ofte, mens ansikts-og nakkemuskler ikke påvirkes (4). En asymptomatisk Hyperkemi (5-80 GANGER CPK normale nivåer) hos unge pasienter regnes som et preklinisk stadium av sykdom og kan vedvare i flere år (1, 5). Alderen på muskel svakhet oppstår vanligvis ved 15 år, selv om det kan oppstå tidligere (<12 år) eller senere (>30 år) alder. Sykdomsprogresjonen kan føre til tap av ambulasjon, respirasjonssvikt og redusert lungekapasitet i avanserte stadier (6). Hjerteinvolvering (hjerterytmeforstyrrelser, hjerteledningsforstyrrelser, venstre ventrikulær ejeksjonsdysfunksjon) rapporteres bare sporadisk (6, 7).
diagnosen kalpainopati bekreftes ved påvisning av patogene mutasjoner I CAPN3 (15Q15 .1) (4), som koder for forskjellige alternativt skjøtede transkripsjoner. Transkripsjonen i full lengde uttrykkes imidlertid primært i muskelvev (8). Det kodede proteinet (CAPN3) er medlem av ikke-lysosomale Ca++-avhengige cysteinproteaser-familien. I muskel DELTAR CAPN3 i «sarcomere remodeling», som er avgjørende for muskeltilpasning og vekst som svar på funksjonelle og metabolske krav. Hittil er det beskrevet mer enn 490 patogene mutasjoner gjennom CAPN3, hvorav de fleste er enkeltnukleotidendringer (4). Mutasjoner I CAPN3 har vært assosiert med mitokondrielle abnormiteter, vekstsvikt, økt oksidativt stress og sarkomere disorganisering som til sammen bidrar til å gjøre muskelen ute av stand til å holde belastninger, forårsaker dermed myofiber degenerasjon og muskelsvinn (8). Diagnosen kalpainopati kan være utfordrende på grunn av den genetiske heterogeniteten og ikke-spesifisiteten til klinisk og instrumentelt mønster. Faktisk er fordeling av muskelsvakhet / atrofi, hypertrofi / pseudo-hypertrofi,og sene kontrakturer svært ofte delt med Andre LGMDs eller nevromuskulære lidelser. Muskelbiopsi mønster i calpainopati er generelt ikke-spesifikk også, alt fra milde muskulære abnormiteter til alvorlige dystrofiske forandringer. Videre er immunhistokjemiske / biokjemiske markører vanligvis ikke pålitelige: calpain-signalet kan være normalt selv i nærvær av et ikke-funksjonelt protein, og omvendt kan det reduseres selv i andre muskeldystrofier som er forskjellige fra calpainopati.
det anbefales derfor å utføre en differensialdiagnose for å gi nøyaktige og pålitelige resultater. Til dette formål er det utviklet molekylærgenetiske testmetoder for å bekrefte diagnosen kalpainopati, inkludert multigenpanel, omfattende genomisk (eksom / genomsekvensering)og enkeltgenanalyse (direkte sekvensering) (9). Implementeringen Av Neste Generasjons Sekvensering (Ngs) var nyttig for å generere informative data som skal brukes til diagnostiske, prediktive eller terapeutiske formål (10-12). Ngs-genpaneler er basert på analysen av et sett med gener assosiert med en bestemt sykdom eller en gruppe relaterte lidelser, som er preget av genetisk og fenotypisk heterogenitet. Ngs-paneler kan også være nyttige for å oppdage blandede eller komplekse fenotyper, som skyldes arv av mer enn en genetisk defekt fra foreldrene (13). Generelt er pasienter som presenterer komplekse fenotyper, som er negative for de kjente mutasjonene i spesialdesignede diagnostiske genpaneler og krever en bred differensialdiagnose, kvalifisert for hele eksom-eller genomsekvensering. Hele exome / genome-sekvensering er dyrere tilnærminger når det gjelder datahåndtering, tolkning og analysekostnader, men øker sannsynligheten for å gi en molekylær diagnose til en mistenkt genetisk lidelse (13). Når Det gjelder LGMDs, er dedikerte ngs-paneler sterkt anbefalt for å sikre høye diagnostiske priser, optimal dekning,følsomhet og spesifisitet ved klinisk testing (9). Ngs-paneler representerer derfor et av de beste systemene for å legge til rette for differensialdiagnose, identifisere nye forårsakende mutasjoner og avklare genotype-fenotype korrelasjoner (14, 15).
i dette manuskriptet rapporteres tilfellet av en pasient som er berørt AV LGMD2A, hvor anvendelsen AV ngs-panelet tillot ikke bare å bekrefte diagnosen calpainopati (mutasjoner I CAPN3), men også å identifisere en ekstra ny mutasjon I LMNA-genet assosiert med utvidet kardiomyopati. Gitt disse resultatene ble analysen utvidet til probandens familiemedlemmer for å gi en mer omfattende tolkning av analytiske data i forhold til de patologiske fenotypene.
Sakspresentasjon
Klinisk Karakterisering Av Proband Og Slektninger
sykdomsutbruddet i probanden ble henvist ved 10 års alder, da hun rapporterer tilstedeværelse av kalvhypertrofi med tuppet å gå, vanskeligheter med å løpe, klatre trapper og stå opp. Symptomene viste en langsom, men progressiv forverring, med påfølgende involvering av proksimale øvre lemmer. I en alder av 13 ble høye NIVÅER AV CPK (~8,000 UI/L) oppdaget, aldri forbundet med myoglobinuri. Suksessivt gjennomgikk pasienten flere nevromuskulære undersøkelser på ulike spesialiserte sentre. To muskelbiopsier ble utført, som viser klassisk dystrofisk bilde (hypertrofiske/atrofiske fibre, indre kjerner, nekrotiske fibre, økning i bindevev) med ikke-spesifikke egenskaper. Som forventet var immunokjemi-og immunoblotstudier ikke entydige som indikerte unormale / reduserte hryvnias, β, γ sarkoglykan, β-dystroglycan og dystrofinsignal kun i nekrotiske fibre (gjentatt immunokjemi resulterte normalt, også for laminin); immunobloter for dystrofin og calpain viste normale signaler. Imidlertid er calpain – 3 immunoblot kjent for å være ikke-helt følsom for lgmd2a diagnose, siden 20-30% av tilfellene viser en normal mengde protein (1, 16-18). På grunn av den kliniske presentasjonen (hyperckmi, kalvhypertrofi/pseudo-hypertrofi) ble dystrofinopatier først utelukket. I tillegg viste ANALYSE AV DMD (Xp21), FKRP (19q13.32) og DYSF (2P13.2) gener ikke patogene mutasjoner. Pasient kom til vår observasjon i en alder av 30, presentere aksial og belte engasjement både i øvre og nedre lem, med betydelig vaggende gangart og bevingede scapulae. I nedre lemmer var svakhet fremtredende i quadriceps og glutei muskler som var signifikant ipo-atrofisk, sammen med bevis på» tilsynelatende hypertrofisk » (pseudo-hypertrofi) kalvemuskler (Figur 1). Kramper, myalgi og rippling ble ikke observert. Komplett pneumologisk (spirometri og polysomnografi) og kardiologisk (ekkografi, 24-H EKG Holter-overvåking, hjerte MR, komplett kardiologisk fysisk inspeksjon med målrettet medisinsk historie og kardiovaskulær refleksanalyse) undersøkelser avslørte ikke noen signifikant abnormitet. Vi utelukket også syremaltasemangel (normale enzymaktivitetstester i lymfocytter/leukocytter). Muskelmagnetisk Resonans Imaging (MRI) viste en fremtredende involvering av scapular belte, paravertebrale muskler, bakre kammer muskler i låret (med relativ sparing av sartorius og gracilis muskler) og av benet (spesielt gastrocnemius medialis og soleus) (Figur 2). Dette mønsteret av involvering er allerede rapportert i litteraturen om MR-bilde ved kalpainopati (19, 20). I TILLEGG viste MR-studien at kalvemuskler var effektivt atrofiske og preget av fettinfiltrasjon, noe som forårsaket en muskuløs » utvidelse.»Den kliniske presentasjonen, begynnelsesalderen, progresjonshastigheten, fordelingen av muskel svakhet og MR-funnene i proband var helt i samsvar med en diagnose AV LGMD2A som er omfattende beskrevet i litteraturen (4, 21, 22).
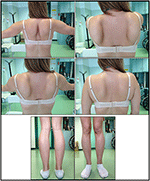
Figur 1. Proband kliniske egenskaper: winged scapulae og kombinasjon av låratrofi og kalv pseudo-hypertrofi.
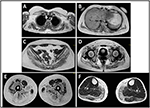
Figur 2. Proband muskel MR: involvering av øvre lem belte og paravertebrale muskler (A, B), involvering av nedre lem belte og bakre kammer av låret (C–E, for det meste glutei) med relativ sparing av sartorius og gracilis, og involvering av bakre kammer av benet (F, for det meste gastrocnemius medialis / soleus).
evalueringen av familiemedlemmer av proband avslørte ikke noen historie med nevromuskulær sykdom og ingen tegn på slektskap mellom foreldrene. Imidlertid presenterte moren til probanden i en alder av 55 år 2 synkopale episoder som hun ble diagnostisert med idiopatisk bradykardi (opptil 40 bpm om natten). Omfattende kardiologiske undersøkelser på probanens mor diagnostiserte en symptomatisk atrioventrikulær (AV) blokk etter forekomst av flere synkopale episoder / lipotymi. Analysen viste tilstedeværelsen av en grunnleggende FØRSTE-graders av-blokk, med flere perioder med alvorlig bradykardi, for det meste nattlig og ofte symptomatisk, assosiert med noen faser av annengrads av-blokk, både type 1 og 2, og noen faser av nattlig fullstendig av dissosiasjon. Ekkokardiografi og ergometrisk test viste ingen signifikante endringer, unntatt for patent foramen ovale. Moren hadde ingen andre kliniske symptomer, predisponerende tilstander eller risikofaktorer. Gitt hennes kliniske bilde ble moren til probanden implantert MED PMK. I tillegg avslørte familiehistorie at bestemor og bestefar av probanden også ble implantert med PMK i henholdsvis 55 og 30 år. Videre døde broren til probandens bestemor ved 51 for alvorlig utvidet kardiomyopati. Kardiologiske undersøkelser ble også utført på far og morbror av proband som resulterte helt upåvirket.
denne studien ble godkjent Av etikkomiteen I Santa Lucia Foundation og ble utført i henhold til Helsinkideklarasjonen. Alle deltakerne ga signert informert samtykke til genetisk analyse, og i denne forbindelse ga de også samtykke til publisering av denne saksrapporten.
Laboratorieundersøkelser Og Diagnostiske Tester
Genomisk DNA ble ekstrahert fra perifert blod (400 µ) ved Hjelp Av MagPurix Blod-DNA-Ekstraksjonssett og MagPurix Automatisk Ekstraksjonssystem (Resnova) i henhold til produsentens instruksjoner. Prøvene ble sekvensert Ved Hjelp Av Ion PGM System Og Ion Ampliseq Tilpasset Panel Høy Spesifisitet (Thermo Fisher Scientific). Størrelsen på panelet var 129.13 Kb, som forventes å skjerme ~99.72% av det totale panelet med en minimumsdekning PÅ 20x. panelet inkluderte 18 gener, som ble valgt ved hjelp av vitenskapelig litteratur, GeneReviews (www.ncbi.nlm.nih.gov/books/NBK1116/) og frekvens av patogene varianter i den generelle populasjonen. En detaljert beskrivelse AV ngs-panelet er oppsummert I Tabell 1.
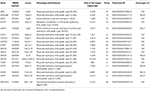
Tabell 1. Tilpasset NGS panel benyttes FOR lgmd diagnose.
Biblioteker byggingen ble utført Av Ion Amliseq™ Bibliotek Kits 2.0. Omtrent 10 ng / µ med start-DNA ble benyttet for multiplex PCR-reaksjoner. Suksessivt ble to rensingstrinn (Ved Hjelp Av AMPure XP, Beckman Coulter) utført for å fjerne uønskede forurensninger og en endelig PCR ble utført. Mal forsterkning og berikelse trinnene ble utført Av Ion Pgm Hi-Q OT2 kit-400, Ion OneTouch 2 System Og Ion OneTouch ES (Thermo Fisher Scientific). Prøvene ble behandlet Av Ion PGM Hi-Q Sekvensering Kit (400 bp, Thermo Fisher Scientific) og kjøre På Ion 316 Chip v2 (850 strømmer kreves) Og Ion PGM Sequencer (Thermo Fisher Scientific). Resultatene ble analysert Ved Hjelp Av Ion Reporter 4.6 (Thermo Fisher Scientific) og Integrated Genome Viewer (IGV). Tolkningen av genetiske varianter ble utført Av Human Gene Mutation Database (HGMD), Leiden Open Variation Database (LOVD), ClinVar og ExAC. Den funksjonelle effekten av de påviste variantene ble evaluert av bioinformatiske prediktive verktøy, inkludert Mutation Taster, Varsome, SIFT, PolyPhen 2, SMART, Human Spleising Finder (HSF). Direkte sekvensering (BigDye Terminator v3.1, BigDyeX Terminator OG ABI3130, Anvendt Biosystems) ble utført for å bekrefte genetiske varianter og sekvens genomisk koding regioner med en dekning <20X (Lmna, Chr1:156106052-156106076; DYSF, Chr2:71753352-71753502, Chr2:71776385-71776640; SGCB, Chr4:52904225-52904560; sgca, chr17:48243242-48243570).
Resultater
ngs-analysen av probanden avslørte 3 heterozygote varianter (Supplerende Figur 1). To varianter ble lokalisert I CAPN3, NEMLIG NM_000070. 2 (CAPN3): c.550dela (S.Thr184Argfs) og c.1813g>C (S.Val605Leu) i henholdsvis exons 4 og 16. Den første er en enkelt nukleotid sletting og er den vanligste (75% av tilfellene) patogene varianten I Europeiske Land (1). Som forventet klassifiserte den bioinformatiske analysen c.550delA (rs80338800) som en funksjonstapvariant (S.Thr184Argfs), noe som forårsaket en rammeskift av den åpne leserammen. De prediktive verktøyene (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) beskrev varianten som skadelig. I TILLEGG VISTE SMART at det endrede proteinproduktet mangler calpain 3-domenet og de tre «EF-hands» – motivene. Det første domenet er involvert I Signalveiene Til Calpain, MENS EF-hendene er avgjørende for Ca++ – avhengig aktivering av proteinet (4).
Om c.1813g> C, er det en roman missense variant (p.Val605Leu) som har blitt spådd å være skadelig (Ved Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) på grunn av en potensiell endring av skjøting. Denne varianten er ikke merket i litteratur eller i nettdatabaser, og den er ikke funnet i 200 kontrollfag. Dessverre ga analysen AV SMART tool ikke betydelige resultater, da varianten ligger innenfor et ukarakterisert domene. Segregeringsanalysen AV CAPN3 viste at moren og morbroren var begge heterozygote for c.550dela, mens faren var bærer av c. 1813g > C.
I tillegg viste ngs-analyse på probanden tilstedeværelsen av en ny variant I LMNA, nemlig NM_170707 (LMNA): c.550c > T (p.Gln184*). Den over nevnte varianten har blitt spådd å være en nullvariant, har ennå ikke blitt beskrevet i litteratur eller blant nettbaserte databaser, og den har ikke blitt funnet i 200 kontrollpersoner. Prediktive verktøy (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) beskrev en patogen effekt av denne varianten på proteinproduktet. SMART tool rapporterte at det endrede lmna-proteinet mangler filamentdomenet, noe som er viktig for å opprettholde sin struktur og funksjon. Segregeringsanalysen fremhevet tilstedeværelsen av c.550c >t-variant bare hos moren, noe som tyder på en mulig sammenheng med hennes hjertesymptomer og hennes familiehistorie av sykdom.
i henhold til kriteriene fastsatt Av American College Of Medical Genetics (ACMG) Standarder Og Retningslinjer (23), kan c.1813g > C (CAPN3) beskrives som en sannsynlig patogen variant med tanke på at DEN ligger i et kritisk domene uten godartet variasjon (PM1); den er fraværende I ExAc, GnomAD og 1000 Genome Browser databaser (PM2); med tanke på recessiv arvemodell har den blitt påvist i trans med en patogen variant (PM3); flere linjer med beregningsbevis støtter en skadelig effekt på genet eller genproduktet (PP3). Når det gjelder den kliniske klassifiseringen av c. 550C>T (LMNA) VED ACMG, kan DEN betegnes som en patogen variant, siden det er en nullvariant som forårsaker tap av funksjon (P.Gln184*) I LMNA (PVS1); den er fraværende I ExAc, GnomAD OG 1000 Genom Browser databaser (PM2); den ligger i et kritisk domene uten godartet variasjon (PM1).; flere linjer med beregningsbevis støtter en skadelig effekt på genet eller genproduktet (PP3).
Konklusjoner
denne saksrapporten presenterte en pasient påvirket AV LGMD, som ble funnet å være bærer av mutasjoner, ikke bare I CAPN3 (c.550dela og c.1813g> C), men også I LMNA (c.550c>T). Disse resultatene forklarer probandens nevromuskulære fenotype på GRUNN AV CAPN3-mutasjoner og fremhever en potensiell risiko for kardiovaskulære lidelser på grunn AV tilstedeværelsen AV varianten I LMNA og hennes positive familiehistorie. Gitt disse resultatene ble familiemedlemmene utsatt for segregeringsanalysen. Spesielt resulterte moren til Å være bærer Av Henholdsvis CAPN3_c.550dela og lmna_c. 550c>T. Moren kan betraktes som en sunn bærer For CAPN3_c. 550delA patogen mutasjon assosiert med calpainopati. Tilstedeværelsen av en ny variant i LMNA kan imidlertid forklare hennes kardiovaskulære patologi (bradykardi og synkopale episoder). Fraværet av nevromuskulær symptomatologi hos moren og det særegne kliniske bildet av proband utelukket en mulig forening Av LMNA_c.550C> T med nevromuskulær fenotype, spesielt Angående Emery-Dreifuss Muskeldystrofi (EMD). FAKTISK påvirker EMD spesielt vastus lateralis og biceps brachii muskler som er relativt spart I LGM2A (24, 25).
mors onkel var heterozygot bare For CAPN3_c. 550delA, og viste som forventet ingen nevromuskulære eller kardiovaskulære problemer. Faren resulterte i å bære romanen CAPN3_c.1813g> C OG dermed var han upåvirket. Samlet sett bekreftet segregeringsanalysen arven til de tre mutasjonene i probanden fra hennes slektninger og fremhevet en kjennskap til kardiomyopati som ikke kan overses (Figur 3).
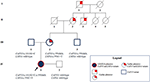
Figur 3. Stamtavle som viser positiv kjennskap til kardial fenotype arvet av mors avstamning og overføring AV CAPN3-og LMNA-varianter i hele familien.
Til sammen gir disse dataene noen viktige hensyn. FOR DET første var ngs-panelet i denne pasienten kritisk for å nå den molekylære diagnosen kalpainopati, oppdage en kjent mutasjon og en annen ny variant spådd som patogen. Sekund, ngs panel tillatt identifisering av romanen LMNA_c. 550C> T variant i proband. Dette resultatet var i samsvar med den positive familiehistorien og med segregeringsdata, forklarer kardiovaskulær sykdom hos moren og, enda viktigere, anbefale mer spesifikk kardiologisk oppfølging i proband. Det er viktig å merke seg at kardiologiske manifestasjoner skjedde i moren senere i alderen (55 år), slik at vi kan anta at probanden fortsatt kan være i en «asymptomatisk kardiologisk fase.»Et variabelt fenotypisk uttrykk FOR LMNA-mutasjon i proband sammenlignet med moren kan imidlertid ikke utelukkes. Alle disse dataene understreker viktigheten av en integrert tilnærming mellom klinikere og genetikere, for korrekt tolkning av resultater, riktig genetisk rådgivning og til slutt klinisk styring og oppfølging. Med hensyn til genetisk og familiær rådgivning ble ngs-analysen også utført på probandens partner for å estimere reproduktiv risiko for paret. Partneren var negativ til alle de 18 testede genene, noe som betyr at han har 1/650 gjenværende risiko for å være en sunn bærer FOR lgmd-forårsakende mutasjoner, med tanke på at testen er 84% følsom. Med tanke på probandens genetiske profil og følsomheten TIL ngs-testen, er gjenværende risiko for paret å få et barn berørt av calpainopati 1/1300. PÅ den annen side overføres lmna patogene varianter i henhold til et autosomalt dominant mønster, noe som betyr at probanden har 50% sannsynlighet for å ha heterozygote barn. Imidlertid kan det kliniske bildet av avkommet ikke sikkert forutsies Da LMNA_c. 550C>T ER en ny variant, og dens funksjonelle innvirkning på fenotypen er fortsatt ukjent.
avslutningsvis fremhever denne saksrapporten den kliniske nytten AV ngs-paneler for å gi nøyaktig lgmd2a-diagnose og beskrive komplekse fenotyper og komorbiditeter som stammer fra arv av forskjellige mutasjoner i flere gener. ANVENDELSEN AV NGS i klinisk praksis bør imidlertid alltid kombineres med en pre-og postgenetisk rådgivning for å gi en klar forklaring på resultatene, mulige implikasjoner på pasientens fenotype, tilbakefall risiko i familien samt å forklare mulige uventede funn.
Datatilgjengelighet
Ingen datasett ble generert eller analysert for denne studien.
Ethics Statement
denne studien ble utført I samsvar Med anbefalingene fra ethics committee Of Santa Lucia Foundation med skriftlig informert samtykke fra alle fag. Alle fagene ga skriftlig informert samtykke i samsvar Med Helsinkideklarasjonen. Protokollen ble godkjent Av ethics committee Of Santa Lucia Foundation.
Forfatterbidrag
RC, CSt, VC, GC, RG, GPa, SC, CP og JM bidro til innsamling av data, analyse og tolkning av data. CSt, RC, GC, RG, GM, OG EG har vært involvert i utarbeidelsen av manuskriptet. SZ, GPr, CSa og SS har vært involvert i innhenting av kliniske data. RC, CSt, VC, GC, RG, GPA, SC, CP, JM, SZ, GPr, GM, CSa, SS og EG har gitt endelig godkjenning av versjonen som skal publiseres.
Finansiering
dette arbeidet støttes AV 5x 2016 National Health Ministry 2.
Interessekonflikt
forfatterne erklærer at forskningen ble utført i fravær av kommersielle eller økonomiske forhold som kan tolkes som en potensiell interessekonflikt.
Supplerende Materiale
Supplerende Materiale for denne artikkelen kan bli funnet online på: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. Protein og genetisk diagnose av lembelte muskeldystrofi TYPE 2a: utbyttet og fallgruvene. Muskel Nerve. (2015) 52:163–73. doi: 10.1002 / mus.24682
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
2. Nigro V, Savarese M. Genetisk grunnlag for lemmer-belte muskeldystrofier: 2014-oppdateringen. Acta Myol. (2014) 33:1–12.
PubMed Abstract | Google Scholar
3. Richard I, Hogrel JY, Stockholm D, Payan CAM, Fougerousse F, Eymard B, et al. Natural history OF LGMD2A for avgrense utfallsmål i kliniske studier. Ann Clin Transl Neurol. (2016) 3:248–65. doi: 10.1002 / acn3.287
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
4. Angelini C, Fanin M. Calpainopathy. In: Editors.editors. editors. editors. GeneReviews® Universitetet I Oslo (2017) 1-30.
PubMed Abstract | Google Scholar
5. C, Angelini C, Schaefer J, Sacconi S, Siciliano G, Vilchez JJ, et al. EFNS retningslinjer for diagnostisk tilnærming til pauci – eller asymptomatisk hyperkemi. Eur J Neurol. (2010) 17:767–73. doi: 10.1111 / j.1468-1331. 2010. 03012.x
Kryssref Fulltekst | Google Scholar
6. Mori-Yoshimura M, Segawa K, Minami N, Oya Y, Komaki h, Nonaka I, et al. Kardiopulmonal dysfunksjon hos pasienter MED lembelte muskeldystrofi 2A. Muskel Nerve. (2017) 55:465–9. doi: 10.1002 / mus.25369
CrossRef Fulltekst | Google Scholar
7. Okere A, Reddy SS, Gupta S, Shinnar M. en kardiomyopati hos en pasient med lembelte muskeldystrofi TYPE 2a. Circ Hjerte Mislykkes. (2013) 6: e12–13. doi: 10.1161 / CIRCHEARTFAILURE.112.971424
Kryssref Fulltekst | Google Scholar
8. Kramerova I, Ermolova N, Eskin A, Hevener A, Quehenberger O, Armando AM, et al. Manglende oppregulering av transkripsjon av gener som er nødvendige for muskeltilpasning, ligger til grunn for lembelte muskeldystrofi 2A (calpainopati). Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093/hmg | ddw086
PubMed Abstrakt | CrossRef Fulltekst / Google Scholar
9. Thompson R, Straub V. Lembelte muskeldystrofier-internasjonale samarbeid for translasjonsforskning. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038 / nrneurol.2016.35
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
10. C, Caputo V, Galota MR, Zampatti S, Marella G, Mauriello S, et al. Anvendelse av presisjonsmedisin i nevrodegenerative sykdommer. Front Neurol. (2018) 9:701. doi: 10.3389 / fneur.2018.00701
PubMed Abstract | CrossRef Full Text | Google Scholar
11. C, strafella C, Caputo V, Errichiello V, Zampatti S, Milano F, et al. Mot anvendelse av presisjonsmedisin i Aldersrelatert Makuladegenerasjon. Prog Retin Øye Res. (2017) 63: 132-46. doi: 10.1016 / j. preteyeres.2017.11.004
PubMed Abstract | CrossRef Full Text | Google Scholar
12. G, g, G, G, G, G, G, G, G, G, G, G, G, et al. Vurdere individuell risiko FOR AMD med genetisk rådgivning, familiehistorie og genetisk testing. Øyne. (2017) 32:446–50. doi: 10.1038 / oye.2017.192
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
13. Adams DR, Eng CM. Neste generasjons sekvensering for å diagnostisere mistenkte genetiske lidelser. N Engl J Med. (2019) 379:1353–62. doi: 10.1056 / NEJMc1814955
PubMed Abstrakt | Kryssref Full Tekst | Google Scholar
14. Angelini C. Nevromuskulær sykdom. Diagnose og oppdagelse i lem-belte muskeldystrofi. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038 / nrneurol.2015.230
CrossRef Fulltekst | Google Scholar
15. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, et al. Bruk av hel-exome sekvensering for diagnose av lem-belte muskeldystrofi: utfall og erfaringer. JAMA Neurol. (2015) 72:1424–32. doi: 10.1001 / jamaneurol.2015.2274
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
16. Milic A, Daniele N, Lochmü H, Mora M, Comi GP, Moggio M, et al. En tredjedel AV lgmd2a biopsier har normal calpain 3 proteolytisk aktivitet som bestemmes av en in vitro-analyse. Neuromusc Disord. (2007) 17:148–56. doi: 10.1016 / j.nmd.2006.11.001
PubMed Abstract | CrossRef Full Text | Google Scholar
17. C, C, C, C, C, C, C, C, C, c, c, et al. Tidlig innsettende calpainopati med normalt ikke-funksjonelt calpain 3-nivå. Dev Med Child Neurol. (2006) 48:304–6. doi: 10.1017 / S001216220600065X
PubMed Abstrakt | Kryssref Fulltekst | Google Scholar
18. Talim B, Ognibene A, Mattioli E, Richard I, Anderson LV, Merlini L. Normal calpain uttrykk i genetisk bekreftet lem-belte muskeldystrofi TYPE 2A.Nevrologi. (2001) 56:692–3. doi: 10.1212 / wnl.56.5.692-a
CrossRef Fulltekst / Google Scholar
19. Díaz-Manera J, Llauger J, Gallardo E, Illa I. MUSKEL-MR i muskeldystrofier. Acta Myol. (2015) 34:95–108.
PubMed Abstract | Google Scholar
20. Mercuri E, Bushby K, Ricci E, Birchall DR, Pane M, Kinali M, et al. Mri-funn i muskler hos pasienter med muskeldystrofi i lemmer med calpain 3-mangel (lgmd2a) og tidlige kontrakturer. Neuromusc Disord. (2005) 15:164–71. doi: 10.1016 / j.nmd.2004.10.008
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Magri F, Nigro V, Angelini C, Mongini T, Mora M, Moroni I, et al. Den italienske lem girdle muskeldystrofi register: relativ frekvens, kliniske egenskaper og differensialdiagnose. Muskel Nerve. (2017) 55:55–68. doi: 10.1002 / mus.25192
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
22. Chae J, Minami N, Jin Y, Nakagawa M, Murayama K, Igarashi F, et al. Calpain 3 genmutasjoner: genetiske og klinisk-patologiske funn i lem-belte muskeldystrofi. Neuromusc Disord. (2001) 11:547–55. doi: 10.1016 / S0960-8966(01)00197-3
PubMed Abstrakt / Fulltekst / Google Scholar
23. Bick D, Das S, Bick D, Bick D, Bick D, Bick D, Bick D, Et al. Standarder og retningslinjer for tolkning av sekvensvarianter: en felles konsensusanbefaling fra American College Of Medical Genetics and Genomics og Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038 / gim.2015.30
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
24. Bonne G, Mercuri E, Muchir A, Urtizberea A, B Hryvcane Hm, Recan D, Et al. Klinisk og molekylær genetisk spektrum av autosomal dominant Emery-Dreifuss muskeldystrofi på grunn av mutasjoner av lamin A / c-genet. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170::HJELP-ANA6>3.0.CO; 2-J
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
25. Hong JS, Ki CS, Kim JW, Suh YL, Kim JS, Baek KK, et al. Hjertedysrytmier, kardiomyopati og muskeldystrofi hos pasienter med Emery-Dreifuss muskeldystrofi og lem-belte muskeldystrofi TYPE 1b.j koreansk Med Sci. (2005) 20:283–90. doi: 10.3346 / jkms.2005.20.2.283
CrossRef Fulltekst / Google Scholar