til tross for den dramatiske nedgangen i humant immunsviktvirus type 1 (HIV-1)-relatert morbiditet og mortalitet etter oppdagelsen av proteasehemmerne og adventen av KOMBINASJONSBEHANDLING med høyaktiv antiretroviral (ARV) på midten av 1990-tallet, mislyktes mange pasienter fortsatt på grunn av resistens og/eller intolerabilitet (1). Det var klart at Flere ARVs som virker på forskjellige trinn i virusets livssyklus, aktiv mot resistente virus og bedre tolerert var nødvendig. Demonstrasjonen av nøkkelrollen til kjemokin-reseptorene CCR5 og CXCR4 i HIV-1-oppføring vekket interesse for denne prosessen som et nytt ARV-mål (2, 3). CCR5 er co-reseptoren for de FLESTE HIV-1-stammer, og disse virusene kalles CCR5 tropic (R5). Virusstammer som bruker CXCR4 kalles CXCR4-tropic (X4), mens stammer som kan bruke begge reseptorene er dual-tropic (4). Virus fra en pasient kan ofte inneholde blandinger Av R5, X4 og dual-tropic stammer, kollektivt kalt CXCR4-bruk.
NØKKELROLLEN TIL CCR5 i HIV-1-oppføring, kombinert med demonstrasjonen at personer som var homozygote for en 32 basepar delesjon I CCR5-genet (CCR5Δ32), og deretter ikke uttrykker funksjonell CCR5, var svært beskyttet mot infeksjon Med R5 HIV-1, fokuserte oppmerksomheten PÅ CCR5 som et attraktivt mål (5). Selv om noen studier har vist subtile effekter AV CCR5Δ 32-mutasjonen på immunfunksjon, som redusert inflammatorisk score hos hepatitt C-infiserte individer og gjenoppretting fra hepatitt b i heterozygoter; mens homozygoter er mer utsatt for kryssbåren encefalitt og alvorlig West Nile virus sykdom, har disse individene liten tilsynelatende bivirkninger på helsen deres (5, 6). Dette, sammen med det faktum at medlemmer Av G-proteinkoblet reseptor superfamily ofte kan knyttes til utvikling av potente, selektive og oralt biotilgjengelige legemidler (7), førte til initiering AV CCR5 ligand-oppdagelsesprogrammer av flere grupper, inkludert et team Fra Pfizer Global Research and Development basert På Sandwich laboratories i Storbritannia.
Maraviroc (UK-427 857, MVC) ble oppdaget ved høy gjennomstrømmingsscreening Av Pfizer compound library ved bruk av en chemokin radioligandbindende analyse. Den mest lovende forbindelsen fra screeningsprosessen ble optimalisert for potens mot reseptoren, antiviral aktivitet, farmakokinetiske egenskaper og selektivitet mot humane cellulære mål gjennom en stor medisinsk kjemiinnsats der nesten 1000 molekyler ble karakterisert (7). MVC binder seg i transmembranlommen TIL CCR5 og er en sakte offset funksjonell antagonist som forhindrer internalisering (7, 8). Den har potent antiviral aktivitet mot ET bredt SPEKTER AV HIV – 1-isolater (7). Sammen med sin utmerkede prekliniske sikkerhetsprofil og akseptable farmakokinetikk, resulterte dette i at den ble nominert som klinisk kandidat i desember 2000 (7).
det var alltid klart at den kliniske utviklingen AV CCR5-antagonister ville være utfordrende, da disse ville være de første vertsmålrettede ARV-legemidlene, og vi ventet derfor inn i ukjent territorium. For å forhindre viktige problemer ble et klinisk utviklingsteam etablert kort tid etter starten av discovery-programmet, og jeg ble rekruttert til å lede det tidlige utviklingsteamet, og begynte I Pfizer i februar 1999. Vi identifiserte flere viktige utfordringer å ta opp i utformingen av det kliniske programmet, i tillegg til å demonstrere sikkerhet og effekt. Den første av disse var at ingen kommersielt tilgjengelig, klinisk validert analyse for å identifisere pasienter infisert Med R5 HIV-1 eksisterte. DETTE var kritisk, DA MVC bare er aktiv mot R5 HIV-1-stammer (7). For det andre, til tross for den tilsynelatende sunne fenotypen til personer MED CCR5Δ 32 (5, 6), var det fortsatt bekymring for sikkerheten ved langvarig eksponering FOR CCR5-antagonister, da blokkering av CCR5 kan være forskjellig fra medfødt fravær AV CCR5-reseptoren, hvor immunsystemet har modnet i fravær AV CCR5 og kompenserende mekanismer kan ha utviklet seg. Til SLUTT, HOS HIV-1-infiserte individer, øker forekomsten AV CXCR4-ved BRUK AV HIV-1-stammer med sykdomsprogresjon og reduksjon I CD4 – celletall (9), selv om det ikke er påvist årsakssammenheng MELLOM cxcr4-ved bruk av virus og CD4-celledeplesjon. Dette har ført til bekymringer for at selektivt trykk fra EN CCR5-antagonist kan drive viruspopulasjonen til å bruke CXCR4 og resultere i CD4-celleforfall.
Fase 1 enkelt-og flerdosestudier med friske frivillige, utført i 2001 og første halvdel av 2002, viste at MVC var sikker og godt tolerert i flere doser opp til 300 mg to ganger DAGLIG (BID), hadde en farmakokinetisk profil som var kompatibel med en gang daglig (QD) eller bid oral dosering, kunne kombineres med Andre Arv-Er, og at doser av ≥100 mg bid resulterte i eksponering over det geometriske gjennomsnittet antivirale IC90 in vitro (7, 10). FOR å demonstrere bevis på farmakologi ble ccr5-reseptormetning målt ved bruk av en skreddersydd ex vivo mip-1β internaliseringstest. Doseavhengig metning ble vist, med doser på ≥25 mg PER døgn som resulterte i nær maksimale metningsnivåer, noe som økte den interessante muligheten FOR AT MVC kunne være effektiv i doser så lave som 25 mg PER døgn. Reseptormetning forble høy i flere dager etter seponering, noe som reflekterte langsom forskyvning fra reseptoren in vivo (11).
vi var begge begeistret og oppmuntret av fase 1-dataene og gikk raskt videre til et fase 2a proof of concept-program. HIV-1-infiserte pasienter ble screenet for Forekomst Av R5-virus kun ved bruk av en ny fenotypisk tropismeanalyse (Trofile®, Monogram Biosciences, South San Francisco, CA, USA), og fikk MVC som monoterapi i 10 dager (12). Ccr5-reseptormetning ble målt i denne studien for å evaluere muligheten for å bruke dette som biomarkør for effekt og i terapeutisk overvåking. De etterlengtede dataene levde opp til våre forventninger og viste at doser av ≥100 mg to GANGER DAGLIG resulterte i gjennomsnittlig MAKSIMAL HIV-1 RNA-reduksjon på > 1.5log10 (Figur 1a), med alle pasienter, unntatt en pasient med X4-virus som feilaktig er inkludert, oppnådde EN HIV-1 RNA-reduksjon på minst 1log10 (12). Dette ga oss tillit til at analysen korrekt identifiserte pasienter som sannsynligvis vil reagere PÅ MVC. HIV-1 rna nadir forekom 1-5 dager etter siste dose MED MVC, i samsvar med langvarig reseptormetning som vist i fase 1-studiene (12). For alle doser unntatt 25 mg QD-reseptormetning på > 80% ble observert gjennom hele doseringsperioden. Det var imidlertid ingen sammenheng mellom reduksjon av virusmengde og grad av reseptormetning. Den mest sannsynlige forklaringen på dette er at svært høye nivåer av reseptormetning er nødvendig for antiviral effekt, og den iboende variabiliteten av analysen tillater ikke differensiering i den grad (11, 12).
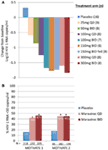
Figur 1. Maraviroc konseptbevis og fase 3 effektresultater. (A) Gjennomsnittlig maksimal endring fra baseline I HIV-1 RNA hos pasienter som får mvc monoterapi. Basert på fase 1-data og modellering og simulering ble doser fra 25 mg DAGLIG TIL 300 mg TO GANGER DAGLIG (inkludert 150 mg to GANGER DAGLIG og fastende) valgt. Hiv-1 rna, sikkerhet og mvc farmakokinetikk ble evaluert (12). (B) MOTIVERE 1 og 2-andel pasienter som oppnår HIV-1 RNA < 50 kopier/mL ved uke 48. HIV-1-infiserte pasienter med R5 HIV-1 og trippelklasseopplevelse og / eller resistens ble randomisert til å få MVC QD ELLER BID, eller placebo, i kombinasjon med et optimalisert antiretroviralt bakgrunnsregime (OBT). P < 0.001 (13, 14).
fase 2a-dataene genererte spenning i hele selskapet, og vi var opptatt av å utvikle det kliniske utviklingsprogrammet så raskt som mulig da det var et høyt medisinsk behov for nye ARVs for å behandle pasienter med ingen eller begrensede behandlingsalternativer. Det omfattende fase 1-programmet (inkludert flere legemiddelinteraksjonsstudier) og et bredt doseområde evaluert i fase 2a proof of concept-studiene, sammen med modellering og simulering, ga oss en meget god forståelse av den sannsynlige effektive dosen AV MVC i kombinasjon med Andre Arv-er. Vi var derfor i stand til å gå direkte til fase 3 effektstudier som evaluerte MVC ved 300 mg (eller tilsvarende, avhengig av samtidig administrerte legemidler) QD og BID, uten å måtte gjøre frittstående fase 2b doseavstandsstudier, og dermed forkorte utviklingstiden betydelig. I slutten av 2004 startet vi fire store studier; MOTIVATE 1 og 2 hos behandlingserfarne pasienter Med R5 HIV-1 (13, 14), MERIT (en fase 3-studie med fase 2b roll-in) i behandling-naï pasienter med R5 HIV-1 (15), og studie A4001029, en fase 2b-sikkerhetsstudie hos behandlingserfarne pasienter med ikke-CCR5 tropisk virus (cxcr4-bruk eller ikke-fenotypbart virus) (16).
dette var en massiv oppgave, med 4794 pasienter screenet på mer enn 200 steder I USA, Canada, Europa, Australia, Sør-Afrika, Mexico og Argentina. To andre SMÅMOLEKYL CCR5-antagonister (aplaviroc og vicriviroc) ble også evaluert i fase 2b-studier på dette tidspunktet (17, 18). I tillegg til de vanlige utfordringene med å håndtere store kliniske studier, ble vi kastet to kurveballer, den første av disse var seponering av aplaviroc på grunn av idiosynkratisk hepatotoksisitet. Det var spekulasjoner om at DETTE kunne være en klasseeffekt AV CCR5-antagonister, DA CCR5-knockout-mus er mer mottakelige concanavalin – en mediert hepatoksisitet (17). I TILLEGG utviklet en pasient i MERIT-studien alvorlig hepatotoksisitet. Dataene antydet at det sannsynligvis var relatert til isoniazid eller kotrimoksazol, men en medvirkende rolle for MVC kunne ikke utelukkes (15). En grundig gjennomgang av alle data for bevis på levertoksisitet FOR MVC og et høyt nivå av årvåkenhet for eventuelle signaler, fant ingen bevis for en systematisk økning i leverenzymer eller andre markører for levertoksisitet. Kort tid etterpå ble det reist bekymringer om en potensiell økt risiko for visse maligniteter, etter forekomst av lymfom hos fire pasienter som fikk vicriviroc i ACTG5211 (18) studie. I utgangspunktet var det bekymringer for at dette kunne være en klasseeffekt basert på IMMUNMODULERENDE potensial FOR CCR5-antagonister, men gjennomgang av data fra andre vicriviroc-studier, samt de pågående MVC-studiene støttet ikke denne teorien (18).
DATA FRA MOTIVATE 1 og 2 og A4001029 var tilgjengelig før VERDIEN, da studiens varighet vanligvis er kortere for studier hos behandlingserfarne pasienter. Det var med stor spenning at vi ventet på uke 24 interimanalyser for MOTIVATE-studiene i oktober 2006, og vi var opprømte over å se at signifikant flere pasienter som fikk MVC hadde ET HIV-1 RNA på <50 kopier/mL (nøkkelmarkøren for effekt) sammenlignet med de som fikk placebo OBT. Dette ble bekreftet av uke 48-dataene, som viste varighet av respons (Figur 1b) (13, 14). I motsetning til dette så pasienter med ikke-CCR5 tropisk HIV-1 som fikk MVC I A4001029 ikke ut til å få signifikant virologisk fordel sammenlignet med placebo (16). Analyse av sikkerhetsdata ga ingen vesentlige bekymringer. Spesielt var det ingen tegn på en negativ effekt på immunfunksjonen, uten økning i episoder med infeksjon eller maligniteter hos MVC-behandlede pasienter. Vurdering av virustropisme ved svikt viste at > 50% av pasientene som ikke fikk MVC-behandling hadde cxcr4-bruk av virus ved svikt, men det var ingen tegn til skadelig effekt PÅ CD4-celletall (14). Virologisk vurdering viste at CXCR4-bruk av viruset som oppsto under mvc selektivt trykk, var fra en eksisterende minoritetsbefolkning og oppsto ikke de novo (19). Samlet sett viste disse resultatene tydelig FORDELENE MED MVC ved behandling av behandlingserfarne pasienter Med R5 HIV-1. En suveren innsats av teamet resulterte i innlevering av saksmapper for registrering i BÅDE USA og Europa bare 2 måneder etter at foreløpige data ble tilgjengelig. MVC (300 mg BID) mottok godkjenning for bruk (i kombinasjon med Andre ARVs) I USA i August 2007, bare 6.5 år etter at den ble nominert som kandidat for klinisk utvikling. En måned senere ble det også godkjent for bruk i DENNE befolkningen I EU.
analysen i UKE 48 av MERIT-studien var skuffende, DA MVC pluss zidovudin/lamivudin (HIV-1 RNA <50 kopier/mL, 65,3%) ikke oppfylte de forhåndsdefinerte kriteriene for ikke-inferioritet (nedre grense for 1-sidig 97,5 konfidensintervall under -10%) til efavirenz pluss zidovudin/lamivudin (HIV-1 RNA <50 kopier/ml, 69,3%) (15). Pasienter for denne studien ble imidlertid screenet For R5-virus ved hjelp av Den opprinnelige Trofile-analysen. Denne analysen har blitt forbedret i mellomtiden for å være mer følsom for påvisning av minoritetspopulasjoner AV cxcr4-bruk av virus. Alle screeningprøver for PASIENTER i MERIT ble deretter testet på nytt ved bruk av enhanced assay og en post hoc-analyse utført kun med pasienter som hadde R5-virus kun ved den mer sensitive assay. I denne analysen var responsraten for MVC og efavirenz henholdsvis 68,3 og 68,5%, med den nedre grensen for 97,5% konfidensintervallet over -10% (15). BASERT på disse dataene ble MVC også godkjent for bruk i behandling-naï pasienter i November 2009 Av United States Food And Drug Administration.
MVC har ikke bare vist seg å være et verdifullt tillegg TIL DET stadig voksende ARV drug armamentarium, men data fra disse studiene har forbedret vår forståelse AV HIV-tropisme og forholdet mellom tropisme og sykdomsprogresjon. For meg personlig representerte dette en periode med stor spenning og tilfredshet, både som lege og forsker.
Forfatterbidrag
ER utarbeidet dette manuskriptet basert på hennes personlige erfaring som medlem AV mvc development team. Alle dataene er publisert i sin helhet andre steder. Alle studier ble utført i samsvar med Prinsippene I Helsinkideklarasjonen og med Alle Internasjonale Konferanse Om Harmonisering God Klinisk Praksis Retningslinjer og lokale regulatoriske og juridiske krav. Alle studier ble godkjent av uavhengige etikkomiteer og alle pasienter ga skriftlig informert samtykke.
Interessekonflikt
Elna Van Der Ryst var ansatt I Pfizer Global Research and development på den tiden MVC ble utviklet. Hun tilbyr for tiden konsulenttjenester Til Pfizer.
Takk
jeg vil gjerne takke kolleger Fra Pfizer som bidro TIL mvc discovery and development program, samt etterforskere og pasienter som deltok i de kliniske studiene.
1. Richman DD, Morton SC, Wrin T, Hellmann N, S Berry, Schapiro MF, et al. Utbredelsen av antiretroviral legemiddelresistens i Usa. AIDS (2004) 18: 1393-401. doi: 10.1097 / 01.hjelp.0000131310.52526.c7
Pubmed Abstrakt | Pubmed Fulltekst | CrossRef Fulltekst | Google Scholar
2. Dr. med. martin SR, Huang Y, Nagashima KA, Et al. HIV-1-inntreden I CD4 + – celler medieres av kjemokin-reseptoren CC-CKR-5. Natur (1996) 381: 667-73. doi:10.1038 / 381667a0
Pubmed Abstrakt | Pubmed Full Tekst | CrossRef Full Tekst | Google Scholar
3. Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry kofaktor: funksjonell cDNA kloning av en syv-transmembran, G protein-koblet reseptor. Vitenskap (1996) 272: 872-7. doi:10.1126 / vitenskap.272.5263.872
CrossRef Fulltekst | Google Scholar
4. Berger EA, Doms RW, Fenyö EM, Korber BT, Littman DR, Moore JP, et al. En ny klassifisering FOR HIV-1. Natur (1998) 391: 240. doi:10.1038/34571
Full Text | Google Scholar
5. Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber C, et al. Resistens MOT HIV – 1-infeksjon hos Kaukasiske Individer som bærer mutante alleler AV CCR5-kjemokin-reseptorgenet. Natur (1996) 382: 722-5. doi:10.1038 / 382722a0
Pubmed Abstrakt | Pubmed Full Tekst | CrossRef Full Tekst | Google Scholar
6. Gilliam BL, Riedel DJ, Redfield RR. Klinisk bruk AV CCR5-hemmere I HIV og utover. J Transl Med (2011) 9 (Suppl 1): S9. doi:10.1186/1479-5876-9-S1-S9
Pubmed Abstrakt | Pubmed Full Tekst | CrossRef Full Tekst | Google Scholar
7. Jørgensen P, Jørgensen M, Jørgensen M, Jørgensen m, et al. Maraviroc (UK-427,857), en potent, oralt biotilgjengelig og selektiv liten molekylhemmer av kjemokin reseptor CCR5 med bredspektret anti-humant immunsviktvirus type 1-aktivitet. Antimikrob Midler Chemother (2005) 49: 4721-32. doi: 10.1128 / AAC.49.11.4721-4732.2005
CrossRef Fulltekst | Google Scholar
8. R, Zhang J, Ji C, Mirzadegan T, Rotstein D, Sankuratri S, et al. Molekylære interaksjoner AV CCR5 med store klasser av små molekyl anti-HIV-1 CCR5 antagonister. Mol Pharmacol (2008) 73: 789-800. doi: 10.1124 / mol.107.042101
Pubmed Abstrakt | Pubmed Fulltekst | CrossRef Fulltekst | Google Scholar
9. Berger EA, Murphy PM, Farber JM. Kjemokinreseptorer som HIV – 1 co-reseptorer: roller i viral oppføring, tropisme og sykdom. Annu Rev Immunol (1999) 17: 657-700. doi:10.1146 / annurev.immunol.17.1.657
Pubmed Abstrakt | Pubmed Fulltekst | CrossRef Fulltekst | Google Scholar
10. Abel S, van Der Ryst E, Rosario MC, James I, Ridgway C, Medhurst C, et al. Vurdering av farmakokinetikk, sikkerhet og toleranse for maraviroc, en NY CCR5-antagonist, hos friske frivillige. Br J Clin Pharmacol (2008) 65 (S1): 5-18. doi:10.1111/j.1365-2125.2008. 03130.x
Pubmed Abstrakt | Pubmed Fulltekst | CrossRef Fulltekst | Google Scholar
11. Rosario MC, Jacqmin P, Dorr P, James I, Jenkins T, Abel S, et al. Farmakokinetisk-farmakodynamisk analyse AV CCR5 – reseptorbelastning av maraviroc hos friske forsøkspersoner og HIV-positive pasienter. Br J Clin Pharmacol (2008) 65 (S1): 86-94. doi:10.1111/j.1365-2125.2008. 03140.x
Pubmed Abstrakt | Pubmed Fulltekst | CrossRef Fulltekst | Google Scholar
12. F hryvnkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AIM, et al. Effekt av kortvarig monoterapi med maraviroc, en NY CCR5-antagonist hos HIV-1-infiserte pasienter. Nat Med (2005) 11: 1170-2. doi:10.1038 / nm1319
Pubmed Abstrakt | Pubmed Fulltekst | CrossRef Fulltekst | Google Scholar
13. Gulick RM, Lalezari J, Goodrich J, Klønete N, DeJesus E, Horban A, et al. Maraviroc for tidligere behandlede pasienter Med R5 HIV-1-infeksjon. N Engl J Med (2008) 359: 1429-41. doi: 10.1056 | NEJMoa0803152
Kryssref Full Tekst / Google Scholar
14. F hryvnkenheuer G, Nelson M, Lazzarin A, Konourina I, Hoepelman IM, Lampiris H, et al. Subgruppeanalyser av maraviroc ved tidligere behandlet R5 HIV-1-infeksjon. N Engl J Med (2008) 359: 1442-55. doi:10.1056 / NEJMoa0803154
Pubmed Abstrakt | Pubmed Full Tekst | CrossRef Full Tekst | Google Scholar
15. A. d., a. d., A. d., A. d., a. d., a. d., a. d., a. d., a. d., a. d., a. d., a. d., et al. Maraviroc versus efavirenz, begge i kombinasjon med zidovudin / lamivudin, til behandling av antiretrovirale-naï personer MED CCR5-tropisk HIV-1. J Infisere Dis (2010) 201: 803-13. doi:10.1086/650697
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst / Google Scholar
16. Saag M, Goodrich J, Fä G, Clotet B, Clumeck N, Sullivan J, Et al. En dobbeltblind, placebokontrollert studie av maraviroc hos behandlingserfarne pasienter infisert med ikke-CCR5-tropisk HIV-1: 24-ukers resultater. J Infisere Dis (2009) 11: 1638-47. doi:10.1086/598965
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst / Google Scholar
17. J. j. w. G., J. J. G., J. J. G., J. j. g., J. j. g., Et al. Levertoksisitet observert i kliniske studier med aplaviroc (GW873140). Antimikrob Agenter Chemother (2008) 52: 858-65. doi: 10.1128 / AAC.00821-07
Pubmed Abstrakt | Pubmed Fulltekst | CrossRef Fulltekst | Google Scholar
18. Chadburn A, Su Z, Henrich TJ, Krambrink A, et al. Lymfomdiagnose og plasma Epstein-Barr-virusbelastning under vicriviroc-behandling: resultater FRA AIDS – kliniske studier gruppe a5211. Clin Infiserer Dette (2009) 48: 642-9. doi:10.1086/597007
Pubmed Abstrakt / Pubmed Fulltekst / CrossRef Fulltekst / Google Scholar
19. Lewis M, Simpson P, Fransen S, Huang W, Whitcomb J, Mosley M, et al. CXCR4-bruk av virus påvist hos pasienter som fikk maraviroc I fase III-studiene MOTIVATE 1 og 2 stammer fra et eksisterende MINDRETALL av cxcr4-brukende virus. Antivir Ther (2007) 12: S65.
Google Scholar