Avec la blessure de tissus sains, une progression prévisible des événements physiologiques se déroule. Cette progression peut être divisée en phases d’inflammation, de prolifération et de maturation. Chaque phase est caractérisée par l’élaboration séquentielle de cytokines distinctives par des cellules spécifiques. Voir les images ci-dessous.
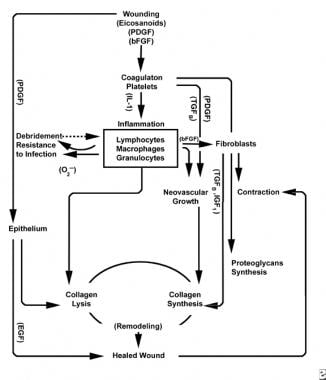 Schémas du processus de cicatrisation.
Schémas du processus de cicatrisation. 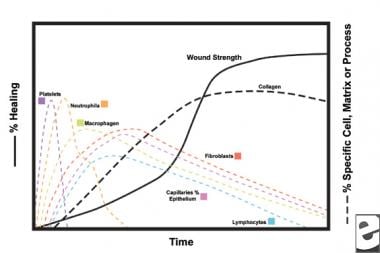 Caractéristiques cellulaires du processus de cicatrisation.
Caractéristiques cellulaires du processus de cicatrisation. La phase inflammatoire
La phase inflammatoire lance simultanément des mécanismes et des voies hémostatiques qui créent les signes cardinaux cliniquement reconnaissables de l’inflammation: rubor (rougeur), calor (chaleur), tumeur (gonflement), dolor (douleur) et functio laesa (perte de fonction).
Une lésion du tissu vasculaire initie la cascade de coagulation extrinsèque en libérant du calcium intracellulaire et du facteur tissulaire qui activent le facteur VII. Le bouchon de fibrine résultant réalise une hémostase assistée par une vasoconstriction réflexe. Ce bouchon agit comme un réseau pour l’agrégation des plaquettes, le type cellulaire le plus courant et le plus « signature » de la phase inflammatoire précoce.
Les plaquettes élaborent un certain nombre de substances pro-inflammatoires, telles que l’adénosine diphosphate, le facteur de croissance tissulaire bêta (TGF-ß) et les facteurs de croissance dérivés des plaquettes (PDGF). Ces facteurs de croissance agissent sur les cellules environnantes et stimulent la chimiotaxie des neutrophiles, des monocytes et des fibroblastes dans la zone de lésion.
Les tissus lésés, par l’intermédiaire de la phospholipase A activée, catalysent simultanément les acides arachidoniques pour produire des prostaglandines vasoactives et des thromboxanes, collectivement appelés eicosanoïdes. Les eicosanoïdes médient l’activité influençant la formation de bouchons plaquettaires, la perméabilité vasculaire et la chimiotaxie cellulaire pour influencer la cicatrisation des plaies. Par exemple, le thromboxane A2 médie la vasoconstriction et l’agrégation plaquettaire.
Après une vasoconstriction initiale, les signes classiques d’inflammation se manifestent par une perméabilité vasculaire accrue. Rubor résulte d’une vasodilatation, médiée par la prostacycline (PGI2), la prostaglandine A (PGA), la prostaglandine D (DPI) et la prostaglandine E (EGP). La tumeur et le calor se développent à mesure que les lacunes endothéliales vasculaires s’agrandissent, permettant la sortie des protéines plasmatiques et du liquide dans l’espace interstitiel. Ces changements sont potentialisés par la PGE2 et la prostaglandine F2a (PGF2a) et permettent l’entrée de cellules inflammatoires dans la zone de lésion, y compris les cellules qui se développent. Dolor est détectée comme PGI2, PGE et PGE2 agissent sur les nocicepteurs périphériques.
Au deuxième stade de la phase inflammatoire, les leucocytes supplantent les plaquettes comme type cellulaire dominant, attirés par la chimiotaxie. Les globules blancs (WBC) sont les cellules prédominantes pendant les 3 premiers jours suivant la blessure; leur nombre atteint son maximum environ 48 heures. Les polymorphonucléocytes (PMN) sont les premiers à commencer des activités bactéricides en utilisant des médiateurs inflammatoires et des métabolites radicalaires sans oxygène. Cependant, une cicatrisation normale peut se produire sans PMNS. Un autre leucocyte, la cellule T auxiliaire, élabore l’interleukine-2 (IL–2). L’IL-2 favorise davantage la prolifération des lymphocytes T pour augmenter la réponse immunogène aux lésions.
Lorsque les leucocytes PMN commencent à décliner après 24 à 36 heures, les monocytes circulants pénètrent dans la plaie et mûrissent dans les macrophages tissulaires. Ces cellules débrident la plaie au niveau microscopique et produisent une grande variété de substances importantes, telles que l’IL-1 et le facteur de croissance basique des fibroblastes (bFGF). L’IL-1 stimule la prolifération des cellules inflammatoires et favorise l’angiogenèse par la réplication des cellules endothéliales. La bFGF est un facteur chimiotactique et mitogène pour les fibroblastes et les cellules endothéliales. Contrairement aux PMN, l’épuisement des macrophages nuit gravement à la cicatrisation des plaies, car le débridement, la prolifération des fibroblastes et l’angiogenèse diminuent tous.
Vers la fin du cycle inflammatoire, le milieu évolutif des eicosanoïdes dans la plaie interagit avec les types cellulaires présents, ce qui entraîne la synthèse de fibroblastes de collagène et de substance broyée (à partir d’un rapport accru de PGF2a à PGE2). De plus, les facteurs de croissance dérivés des macrophages sont maintenant à des niveaux optimaux, influençant fortement l’afflux de fibroblastes, puis de kératinocytes et de cellules endothéliales dans la plaie. Alors que les cellules mononucléées continuent de remplacer les WBC et les macrophages, la phase proliférative commence.
La phase proliférative
Deux à trois jours après la blessure, les fibroblastes migrent vers l’intérieur à partir des marges de la plaie sur la matrice fibrineuse établie pendant la phase inflammatoire. Au cours de la première semaine, les fibroblastes commencent à produire des glycosaminoglycanes et des protéoglycanes, la substance broyée pour le tissu de granulation, ainsi que du collagène, en réponse au bFGF et au TGF-ß synthétisés par des macrophages, ainsi qu’au PDGF.
Les fibroblastes deviennent rapidement le type cellulaire dominant, culminant à 1-2 semaines. Ils génèrent non seulement des molécules de collagène, mais également des cytokines telles que le PDGF, le TGF-ß, le bFGF, le facteur de croissance des kératinocytes et le facteur de croissance insulinique-1. Les fibroblastes assemblent également des molécules de collagène en fibres, qui sont réticulées et organisées en faisceaux. Le collagène est le composant majeur du tissu conjonctif de la plaie aiguë, la production nette se poursuivant pendant les 6 prochaines semaines. La teneur croissante en collagène de la plaie est en corrélation avec l’augmentation de la résistance à la traction.
Les kératinocytes et les cellules endothéliales prolifèrent également pendant cette période, produisant éventuellement des facteurs de croissance autocrines qui maintiennent leur croissance. L’expansion endothéliale contribue à l’angiogenèse, car des vaisseaux intacts génèrent des bourgeons dans le tissu de granulation. La néovascularisation facilite la croissance de la ligne de fibroblastes qui avance dans la plaie, en leur fournissant les nutriments et les cytokines nécessaires.
La dégradation du caillot de fibrine et de la matrice provisoire s’accompagne du dépôt de tissu de granulation (substance broyée, collagène, capillaires), qui se poursuit jusqu’à ce que la plaie soit couverte. La diminution des niveaux d’acide hyaluronique (dans la substance broyée) et l’augmentation des niveaux de sulfate de chondroïtine ralentissent la migration et la prolifération des fibroblastes tout en induisant la différenciation des fibroblastes, passant à la phase de maturation de la cicatrisation.
La phase de maturation
Pendant les 6 premières semaines, la nouvelle production de collagène domine le processus de cicatrisation, déposée de manière aléatoire dans le tissu de granulation aiguë de la plaie. À mesure que la plaie mûrit, le collagène est remodelé en une structure plus organisée avec une résistance à la traction accrue. Progressivement, le collagène de type I remplace le type III jusqu’à ce que le rapport cutané normal de 4: 1 soit atteint. À mesure que le remodelage se poursuit, la collagénolyse de la métalloprotéinase matricielle atteint un état d’équilibre avec la synthèse du collagène. Plateaux de résistance à la traction à 80% de la résistance d’origine environ 1 an après la blessure.
Superficielles à cette activité, les cellules épithéliales continuent de migrer vers l’intérieur à partir du bord de la plaie jusqu’à ce que le défaut soit couvert. À ce stade, l’inhibition de contact induit la transformation des fibroblastes en myofibroblastes, qui contiennent des fibres d’actine contractiles. La contraction de la plaie suit, remplaçant le volume du tissu lésé par un nouveau tissu, bien que le rôle exact du myofibroblaste n’ait pas été complètement élucidé.
Les moyens de dissuasion à la cicatrisation des plaies
Les plaies aiguës passent généralement par un processus de réparation ordonné et rapide qui se traduit par une restauration durable de l’intégrité anatomique et fonctionnelle. Cependant, divers facteurs physiologiques et mécaniques peuvent altérer la réponse de guérison, entraînant une plaie chronique qui ne parvient pas à suivre la progression par étapes habituelle. Les infections locales, l’hypoxie, les traumatismes, les corps étrangers ou les problèmes systémiques tels que le diabète sucré, la malnutrition, l’immunodéficience ou les médicaments sont les plus fréquemment responsables.
Toutes les plaies sont contaminées, mais résistent le mieux aux infections invasives. Lorsque la concentration dépasse 100 000 (105) organismes par gramme de tissu ou que le système immunitaire est compromis, une infection s’ensuit fréquemment. La cellulite prolonge la phase inflammatoire en maintenant des niveaux élevés de cytokines pro-inflammatoires et de protéases tissulaires, qui dégradent les tissus de granulation et les facteurs de croissance tissulaires, et en retardant le dépôt de collagène.
Le débridement (chirurgical, enzymatique et / ou par changement de pansement) et les antibiotiques sont les piliers du traitement antibiotique. Le débridement élimine les tissus dévitalisés, qui peuvent être une source d’endotoxines qui inhibent la migration des fibroblastes et des kératinocytes dans la plaie. Les corps étrangers peuvent également nécessiter un retrait, car la présence d’une suture en soie réduit de 10 000 fois le nombre de bactéries nécessaires pour inciter à l’infection. (Pour une description détaillée de la technique, voir l’article de référence Medscape Sur l’enlèvement de corps étranger de blessure.)
L’hypoxie cellulaire retarde la cicatrisation des plaies par divers moyens. La réticulation des fibrilles de collagène nécessite de l’oxygène pour hydroxyler la proline et la lysine et échoue lorsque la pression tissulaire est inférieure à 40 mm Hg. La puissance bactéricide de la phosphorylation oxydative des leucocytes souffre également dans un environnement hypoxique, réduisant le seuil d’infection. Les mesures visant à améliorer l’apport d’oxygène dépendent de l’étiologie. Le tabagisme, qui provoque une vasoconstriction et augmente l’adhérence plaquettaire, doit être arrêté. Une angioplastie ou un pontage artériel peut être nécessaire pour une maladie vasculaire périphérique. Des mesures d’appoint pour améliorer la perfusion systémique en cas d’insuffisance cardiaque peuvent être indiquées. Une valeur d’hématocrite inférieure à 15% doit être traitée et l’euvolémie rétablie, au besoin. La stase veineuse ou l’insuffisance lymphatique peuvent être améliorées avec des vêtements compressifs.
Une maladie systémique peut prolonger ou interrompre considérablement la cicatrisation des plaies. La glycosylation dans le diabète sucré altère la phagocytose des bactéries neutrophiles et macrophages, prolongeant la phase inflammatoire. La phase proliférative est également prolongée dans la même maladie car les érythrocytes deviennent moins souples et moins capables de fournir de l’oxygène à la plaie pour le métabolisme tissulaire et la synthèse du collagène.
La malnutrition entraîne une diminution de la prolifération des fibroblastes, une altération de la néovascularisation et une diminution de l’immunité cellulaire et humorale. Les plaies exercent des exigences métaboliques accrues, en particulier dans les tissus de granulation. Les acides aminés tels que la méthionine, la proline, la glycine et la lysine sont essentiels au fonctionnement normal des cellules et à la réparation des plaies cutanées. Les acides gras sont des constituants critiques des membranes cellulaires et sont le substrat des eicosanoïdes qui médient le processus inflammatoire. Les acides gras essentiels linolénique et l’acide linoléique doivent être fournis dans l’alimentation, car le corps humain est incapable de synthétiser de novo ces molécules.
Des vitamines et des minéraux adéquats doivent être disponibles pour le métabolisme cellulaire, agissant comme signaux et cofacteurs cellulaires. La vitamine C (acide ascorbique) et le fer sont nécessaires à l’hydroxylation de la lysine et de la proline, qui réticulent et stabilisent la structure en triple hélice du collagène; le cuivre joue également un rôle dans la stabilisation du collagène. La vitamine A (acide rétinoïque) joue un rôle important dans la modulation de la production et de la dégradation du collagène et est particulièrement importante dans l’épithélialisation. Un antioxydant puissant, la vitamine E (alpha-tocophérol) semble accélérer la guérison cutanée et osseuse chez les animaux, et la supplémentation peut avoir un rôle chez les humains. Une carence en oligo-métaux, en particulier en zinc, est également associée à une mauvaise cicatrisation des plaies; celle-ci doit être reconstituée, le cas échéant.
Ovide aurait écrit: « les médicaments guérissent parfois, tuent parfois. »Cela est certainement vrai en ce qui concerne la cicatrisation des plaies. Les corticostéroïdes émoussent les processus de toute la phase inflammatoire. La vitamine A (par voie topique ou 25 000 UI / j par voie orale) atténue les effets curatifs néfastes des corticostéroïdes, mais une hépatotoxicité peut résulter d’une utilisation prolongée (c.-à-d. > 1 mo). Les médicaments anti-inflammatoires non stéroïdiens (AINS) interfèrent également avec le métabolisme de l’acide arachidonique et, par conséquent, la cicatrisation des plaies. De plus, les AINS inhibent la fonction plaquettaire, l’un des premiers processus de la phase inflammatoire.
Une étude de Sutcliffe et al a suggéré que la régulation à la hausse de la protéine connexine de la jonction gap est commune aux plaies chroniques. En examinant la connexine dans trois types de plaies — jambe veineuse, pied diabétique et escarres — les enquêteurs ont constaté que chaque type de plaie présentait une régulation à la hausse de la connexine épidermique 43, de la connexine 26 et de la connexine 30, ainsi que de la connexine cutanée 43.