- Introduction
- Présentation du cas
- Caractérisation clinique du Proband et de ses proches
- Analyses de laboratoire et Tests diagnostiques
- Résultats
- Conclusions
- Disponibilité des données
- Déclaration d’éthique
- Contributions des auteurs
- Financement
- Déclaration sur les conflits d’intérêts
- Matériel supplémentaire
Introduction
Les Dystrophies Musculaires de la ceinture des membres (LGMD) comprennent un groupe hétérogène de troubles caractérisés par l’émaciation progressive et la faiblesse des muscles proximaux de la ceinture des membres (1, 2). Les LGMD présentent une variabilité inter et intrafamiliale, allant de formes très bénignes à des phénotypes sévères, à début précoce et à progression rapide (2). Les LGMD peuvent être classées comme autosomiques dominantes (LGMD1) ou récessives (LGMD2). Le premier groupe a généralement un âge adulte d’apparition et n’est pas très fréquent (< 10% de tous les LGMD), alors que les derniers sont plus fréquents (1:15000) (1). Parmi les formes récessives, la LGMD2A (ou calpainopathie) est la LGMD la plus fréquente dans le monde, touchant environ 30% de tous les cas de LGMD (3). Les signatures cliniques de la maladie comprennent la marche sur la pointe des pieds, la démarche de dandinage, la difficulté à courir et à monter les escaliers, les ailes scapulaires. Des contractures articulaires, un raccourcissement du tendon d’Achille, une scoliose sont couramment observés, tandis que les muscles du visage et du cou ne sont pas affectés (4). Une hypercémie asymptomatique (5 à 80 fois les taux normaux de CPK) chez les jeunes patients est considérée comme un stade préclinique de la maladie et peut persister plusieurs années (1, 5). L’âge d’apparition de la faiblesse musculaire survient généralement à 15 ans, bien qu’il puisse survenir plus tôt (< 12 ans) ou plus tard (> 30 ans). La progression de la maladie peut entraîner une perte de déambulation, une insuffisance respiratoire et une réduction de la capacité vitale pulmonaire aux stades avancés (6). Une atteinte cardiaque (troubles du rythme cardiaque, troubles de la conduction cardiaque, dysfonctionnement de l’éjection ventriculaire gauche) n’est signalée qu’occasionnellement (6, 7).
Le diagnostic de calpainopathie est confirmé par la détection de mutations pathogènes dans CAPN3 (15q15.1) (4), codant différents transcrits alternativement épissés. Cependant, la transcription intégrale est principalement exprimée dans le tissu musculaire (8). La protéine codée (CAPN3) est un membre de la famille des protéases à cystéine non lysosomales dépendantes du Ca++. Dans le muscle, CAPN3 participe au « remodelage du sarcomère », essentiel à l’adaptation et à la croissance musculaires en réponse aux demandes fonctionnelles et métaboliques. À ce jour, plus de 490 mutations pathogènes dans l’ensemble du CAPN3 ont été décrites, dont la plupart sont des modifications mononucléotidiques (4). Des mutations du CAPN3 ont été associées à des anomalies mitochondriales, à une défaillance de la croissance, à une augmentation du stress oxydatif et à une désorganisation des sarcomères qui contribuent à rendre le muscle incapable de supporter les charges, provoquant ainsi une dégénérescence des myofibres et une atrophie musculaire (8). Le diagnostic de calpainopathie peut être difficile en raison de l’hétérogénéité génétique et de la non-spécificité du schéma clinique et instrumental. En effet, la répartition de la faiblesse/atrophie musculaire, de l’hypertrophie/pseudo-hypertrophie, et des contractures tendineuses sont très souvent partagées avec d’autres LGMD ou troubles neuromusculaires. Le schéma de biopsie musculaire dans la calpainopathie est généralement non spécifique, allant d’anomalies musculaires légères à de graves modifications dystrophiques. De plus, les marqueurs immunohistochimiques / biochimiques ne sont généralement pas fiables: le signal de la calpaïne peut être normal même en présence d’une protéine non fonctionnelle, et vice versa, il peut être réduit même dans d’autres dystrophies musculaires différentes de la calpaïnopathie.
Il est donc recommandé d’effectuer un diagnostic différentiel afin de fournir des résultats précis et fiables. À cette fin, des approches de tests génétiques moléculaires ont été développées pour confirmer le diagnostic de calpainopathie, y compris le panel multigénique, l’analyse génomique étendue (séquençage exome/génome) et l’analyse monogénique (séquençage direct) (9). La mise en œuvre du séquençage de nouvelle génération (NGS) a été utile pour générer des données informatives à appliquer à des fins diagnostiques, prédictives ou thérapeutiques (10-12). Les panels de gènes NGS sont basés sur l’analyse d’un ensemble de gènes associés à une maladie spécifique ou à un groupe de troubles apparentés, caractérisés par une hétérogénéité génétique et phénotypique. Les panneaux NGS peuvent également être utiles pour détecter des phénotypes mixtes ou complexes, qui résultent de l’héritage de plus d’un défaut génétique des parents (13). En général, les patients présentant des phénotypes complexes, négatifs aux mutations connues dans des panels de gènes diagnostiques conçus sur mesure et nécessitant un diagnostic différentiel large, sont éligibles au séquençage de l’exome entier ou du génome. Le séquençage de l’exome entier/génome est une approche plus coûteuse en termes de gestion des données, d’interprétation et de coûts analytiques, mais augmente la probabilité de fournir un diagnostic moléculaire à une maladie génétique suspectée (13). Concernant les LGMD, des panels NGS dédiés sont fortement recommandés, afin d’assurer des taux de diagnostic élevés, une couverture optimale, une sensibilité et une spécificité des tests cliniques (9). Les panels NGS représentent donc l’un des meilleurs systèmes pour faciliter le diagnostic différentiel, identifier de nouvelles mutations causales et clarifier les corrélations génotype-phénotype (14, 15).
Dans ce manuscrit, le cas d’un patient atteint de LGMD2A est rapporté, pour lequel l’application du panel NGS a permis non seulement de confirmer le diagnostic de calpainopathie (mutations du CAPN3) mais également d’identifier une mutation supplémentaire et nouvelle du gène LMNA associée à une cardiomyopathie dilatée. Compte tenu de ces résultats, l’analyse a été étendue aux membres de la famille du proband afin de fournir une interprétation plus complète des données analytiques par rapport aux phénotypes pathologiques.
Présentation du cas
Caractérisation clinique du Proband et de ses proches
L’apparition de la maladie dans le proband a été signalée à l’âge de 10 ans, lorsqu’elle a signalé la présence d’une hypertrophie du mollet avec marche sur la pointe des pieds, des difficultés à courir, à monter les escaliers et à se lever. Les symptômes ont montré une aggravation lente mais progressive, suivie d’une atteinte du membre supérieur proximal. À l’âge de 13 ans, des niveaux élevés de CPK (~ 8 000 UI /L) ont été découverts par hasard, jamais associés à une myoglobinurie. Successivement, le patient a subi plusieurs examens neuromusculaires dans divers centres spécialisés. Deux biopsies musculaires ont été réalisées, montrant une image dystrophique classique (fibres hypertrophiques / atrophiques, noyaux internes, fibres nécrotiques, augmentation du tissu conjonctif) avec des caractéristiques non spécifiques. Comme prévu, les études d’immunochimie et d’immunoblot n’ont pas été concluantes indiquant un signal α, β, γ sarcoglycane, β-dystroglycane et dystrophine anormal / réduit uniquement dans les fibres nécrotiques (une immunochimie répétée a entraîné une normale, également pour la laminine); les immunoblots pour la dystrophine et la calpaïne ont révélé des signaux normaux. Cependant, l’immunoblot calpain-3 est connu pour ne pas être complètement sensible au diagnostic LGMD2A, car 20 à 30% des cas présentent une quantité normale de protéines (1, 16-18). En raison de la présentation clinique (hypercmie, hypertrophie du mollet / pseudo-hypertrophie), les dystrophinopathies ont d’abord été exclues. De plus, l’analyse des gènes DMD (Xp21), FKRP (19q13.32) et DYSF (2p13.2) n’a pas révélé de mutations pathogènes. Le patient est venu à notre observation à l’âge de 30 ans, présentant une atteinte axiale et ceinturale à la fois dans les membres supérieurs et inférieurs, avec une démarche de dandinage importante et des omoplates ailées. Dans les membres inférieurs, la faiblesse était importante dans les quadriceps et les muscles fessiers qui étaient significativement ipo-atrophiques, ainsi que la preuve de muscles du mollet « apparemment hypertrophiques » (pseudo-hypertrophie) (Figure 1). Les crampes, les myalgies et les ondulations n’ont pas été observées. Les examens pneumologiques complets (spirométrie et polysomnographie) et cardiologiques (échographie, surveillance Holter ECG 24 h, IRM cardiaque, inspection physique cardiologique complète avec antécédents médicaux ciblés et analyse réflexe cardiovasculaire) n’ont révélé aucune anomalie significative. Nous avons également exclu un déficit en maltase acide (tests d’activité enzymatique normaux dans les lymphocytes / leucocytes). L’imagerie par résonance magnétique musculaire (IRM) a montré une atteinte importante de la ceinture scapulaire, des muscles paravertébraux, des muscles du compartiment postérieur de la cuisse (avec une épargne relative des muscles sartorius et gracilis) et de la jambe (en particulier gastrocnemius medialis et soleus) (Figure 2). Ce schéma d’implication a déjà été rapporté dans la littérature concernant l’image IRM dans la calpainopathie (19, 20). De plus, l’étude IRM a montré que les muscles du mollet étaient effectivement atrophiques et caractérisés par une infiltration de graisse, provoquant un « élargissement » musculaire. »La présentation clinique, l’âge d’apparition, le taux de progression, la distribution de la faiblesse musculaire et les résultats de l’IRM dans le proband étaient pleinement compatibles avec un diagnostic de LGMD2A largement décrit dans la littérature (4, 21, 22).
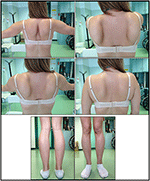
Figure 1. Caractéristiques cliniques proband: omoplates ailées et combinaison d’atrophie de la cuisse et de pseudo-hypertrophie du mollet.
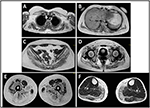
Figure 2. IRM musculaire Proband: atteinte de la ceinture des membres supérieurs et des muscles paravertébraux (A, B), atteinte de la ceinture des membres inférieurs et du compartiment postérieur de la cuisse (C-E, principalement le fessier) avec une relative épargne du sartorius et du gracilis, et atteinte du compartiment postérieur de la jambe (F, principalement gastrocnemius medialis / soleus).
L’évaluation des membres de la famille du proband n’a révélé aucun antécédent de maladie neuromusculaire et aucune preuve de consanguinité entre les parents. Cependant, la mère du proband, à l’âge de 55 ans, a présenté 2 épisodes syncopaux pour lesquels on lui a diagnostiqué une bradycardie idiopathique (jusqu’à 40 bpm la nuit). Des investigations cardiologiques approfondies sur la mère du proband ont diagnostiqué un bloc auriculo-ventriculaire (AV) symptomatique après la survenue de plusieurs épisodes syncopaux / lipothymie. L’analyse a montré la présence d’un bloc AV basique au premier degré, avec plusieurs périodes de bradycardie sévère, principalement nocturne et souvent symptomatique, associées à certaines phases de bloc AV au second degré, de type 1 et 2, et à certaines phases de dissociation AV complète nocturne. L’échocardiographie et le test ergométrique n’ont pas révélé d’altérations significatives, à l’exception du foramen ovale breveté. La mère ne présentait aucun autre symptôme clinique, condition prédisposante ou facteur de risque. Compte tenu de son tableau clinique, la mère du proband a été implantée avec PMK. En outre, les antécédents familiaux ont révélé que la grand-mère et l’arrière-grand-père du proband ont également été implantés avec PMK à l’âge de 55 et 30 ans, respectivement. De plus, le frère de la grand-mère du proband est décédé à 51 ans d’une cardiomyopathie dilatée sévère. Des examens cardiologiques ont également été effectués sur le père et l’oncle maternel du proband qui n’ont pas été affectés.
La présente étude a été approuvée par le comité d’éthique de la Fondation Santa Lucia et a été réalisée conformément à la Déclaration d’Helsinki. Tous les participants ont donné leur consentement éclairé signé pour l’analyse génétique et, à cet égard, ils ont également donné leur consentement pour la publication de ce rapport de cas.
Analyses de laboratoire et Tests diagnostiques
L’ADN génomique a été extrait du sang périphérique (400 µL) à l’aide du Kit d’Extraction d’ADN Sanguin MagPurix et du Système d’Extraction Automatique MagPurix (Resnova) selon les instructions du fabricant. Les échantillons ont été séquencés à l’aide du système PGM Ionique et d’un panneau personnalisé à haute spécificité d’Amplieq Ionique (Thermo Fisher Scientific). La taille du panneau était de 129.13 Kb, qui devrait filtrer ~ 99,72% du panel total avec une couverture minimale de 20X. Le panel comprenait 18 gènes, qui ont été sélectionnés à l’aide de la littérature scientifique, GeneReviews (www.ncbi.nlm.nih.gov/books/NBK1116 /) et la fréquence des variants pathogènes dans la population générale. Une description détaillée du panel NGS est résumée dans le tableau 1.
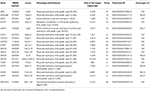
Tableau 1. Panneau NGS personnalisé utilisé pour le diagnostic LGMD.
Bibliothèques La construction a été réalisée par les kits de bibliothèque Ion AmpliSeq™ 2.0. Environ 10 ng/µl d’ADN de départ ont été utilisés pour les réactions de PCR multiplex. Successivement, deux étapes de purification (utilisant AMPure XP, Beckman Coulter) ont été effectuées pour éliminer les contaminants indésirables et une PCR finale a été effectuée. Les étapes d’amplification et d’enrichissement des modèles ont été réalisées par le kit-400 Ion PGM Hi-Q OT2, le système Ion OneTouch 2 et Ion OneTouch ES (Thermo Fisher Scientific). Les échantillons ont été traités par le kit de séquençage Ionique PGM Hi-Q (400 pb, Thermo Fisher Scientific) et exécutés sur puce Ion 316 v2 (850 débits requis) et Séquenceur Ionique PGM (Thermo Fisher Scientific). Les résultats ont été analysés en utilisant Ion Reporter 4.6 (Thermo Fisher Scientific) et Integrated Genome Viewer (IGV). L’interprétation des variants génétiques a été réalisée par la Base de données sur les mutations génétiques Humaines (HGMD), la Base de données Ouverte sur les variations de Leiden (LOVD), ClinVar et ExAC. L’effet fonctionnel des variants détectés a été évalué par des outils prédictifs bioinformatiques, notamment Mutation Taster, Varsome, SIFT, PolyPhen 2, SMART, Human Splicing Finder (HSF). Séquençage direct (Terminateur BigDye v3.1, Terminateur BigDyeX et ABI3130, Applied Biosystems) a été réalisée pour confirmer des variants génétiques et séquencer des régions codantes génomiques avec une couverture < 20X (LMNA, Chr1: 156106052-156106076 ; DYSF, Chr2: 71753352-71753502, Chr2: 71776385-71776640 ; SGCB, Chr4 : 52904225-52904560; SGCA, Chr17: 48243242-48243570).
Résultats
L’analyse NGS du proband a révélé 3 variantes hétérozygotes (Figure supplémentaire 1). Deux variantes ont été localisées dans CAPN3, à savoir NM_000070.2 (CAPN3) : c. 550delA (p. Thr184Argfs) et c. 1813G > C (p. Val605Leu) dans les exons 4 et 16, respectivement. Le premier est une délétion nucléotidique unique et est le variant pathogène le plus courant (75% des cas) dans les pays européens (1). Comme prévu, l’analyse bioinformatique a classé le c.550delA (rs80338800) comme une variante de perte de fonction (p. Thr184Argfs), provoquant un décalage d’image du cadre de lecture ouvert. Les outils prédictifs (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) ont décrit la variante comme délétère. De plus, SMART a révélé que le produit protéique altéré n’avait pas le domaine de la calpaïne 3 et les trois motifs « EF-hands ». Le premier domaine est impliqué dans les voies de signalisation de la Calpaïne tandis que les mains EF sont essentielles à l’activation Ca++-dépendante de la protéine (4).
Concernant c. 1813G >C, il s’agit d’une nouvelle variante de faux sens (p. Val605Leu) dont on a prédit qu’elle serait délétère (par Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) en raison d’une altération potentielle de l’épissage. Cette variante n’est pas annotée dans la littérature ou parmi les bases de données en ligne et elle n’a pas été trouvée chez 200 sujets témoins. Malheureusement, l’analyse par SMART tool n’a pas donné de résultats significatifs, car la variante se situe dans un domaine non caractérisé. L’analyse de ségrégation de CAPN3 a montré que la mère et l’oncle maternel étaient tous deux hétérozygotes pour c. 550delA, tandis que le père était porteur de c. 1813G > C.
De plus, l’analyse NGS sur le proband a révélé la présence d’un nouveau variant dans LMNA, à savoir NM_170707 (LMNA) : c. 550C > T (p. Gln184*). La variante sur-mentionnée a été prédite comme une variante nulle, n’a pas encore été décrite dans la littérature ou parmi les bases de données en ligne et elle n’a pas été trouvée chez 200 sujets témoins. Des outils prédictifs (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) ont décrit un effet pathogène de cette variante sur le produit protéique. SMART tool a rapporté que la protéine LMNA modifiée ne possède pas le domaine du filament, ce qui est essentiel pour maintenir sa structure et sa fonction. L’analyse de la ségrégation a mis en évidence la présence du c.Variante 550C > T uniquement chez la mère, suggérant une association possible avec sa symptomatologie cardiaque et ses antécédents familiaux de maladie.
Selon les critères établis par les normes et directives de l’American College of Medical Genetics (ACMG) (23), c. 1813G > C (CAPN3) peut être décrit comme un variant pathogène probable étant donné qu’il se trouve dans un domaine critique sans variation bénigne (PM1); il est absent des bases de données ExAC, GnomAD et 1000 Genome Browser (PM2).; en considérant le modèle récessif d’hérédité, il a été détecté chez les trans avec un variant pathogène (PM3); plusieurs lignes de données de calcul soutiennent un effet délétère sur le gène ou le produit génétique (PP3). En ce qui concerne la classification clinique de c.550C > T (LMNA) par ACMG, il peut être désigné comme variant pathogène, car il s’agit d’un variant nul entraînant une perte de fonction (p.Gln184*) dans LMNA (PVS1); il est absent des bases de données ExAC, GnomAD et 1000 Genome Browser (PM2); il est situé dans un domaine critique sans variation bénigne (PM1).; plusieurs lignes de données de calcul soutiennent un effet délétère sur le gène ou le produit génétique (PP3).
Conclusions
Ce rapport de cas présentait un patient atteint de MGG, porteur de mutations non seulement dans le CAPN3 (c. 550delA et c. 1813G > C) mais également dans le LMNA (c. 550C > T). Ces résultats expliquent le phénotype neuromusculaire du proband en raison des mutations CAPN3 et mettent en évidence un risque potentiel de troubles cardiovasculaires en raison de la présence du variant dans LMNA et de ses antécédents familiaux positifs. Compte tenu de ces résultats, les membres de la famille ont été soumis à l’analyse d’isolement. En particulier, la mère s’est avérée porteuse de CAPN3_c.550delA et LMNA_c.550C > T, respectivement. La mère peut être considérée comme porteuse saine de la mutation pathogène CAPN3_c.550delA associée à la calpainopathie. Cependant, la présence d’un nouveau variant dans l’ARNMM pourrait expliquer sa pathologie cardiovasculaire (bradycardie et épisodes syncopaux). L’absence de symptomatologie neuromusculaire chez la mère et le tableau clinique particulier du proband excluaient une éventuelle association de LMNA_c.550C > T avec phénotype neuromusculaire, en particulier concernant la dystrophie musculaire d’Emery-Dreifuss (EMD). En fait, l’EMD affecte spécifiquement les muscles vastus lateralis et biceps brachii qui sont relativement épargnés dans LGM2A (24, 25).
L’oncle maternel n’était hétérozygote que pour le CAPN3_c.550delA et, comme prévu, ne présentait aucun problème neuromusculaire ou cardiovasculaire. Le père a eu pour résultat de porter le nouveau CAPN3_c.1813G > C et, par conséquent, il n’a pas été affecté. Dans l’ensemble, l’analyse de ségrégation a confirmé l’héritage des trois mutations du proband de ses proches et a mis en évidence une familiarité avec la cardiomyopathie qui ne peut être négligée (Figure 3).
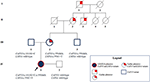
Figure 3. Pedigree montrant la familiarité positive pour le phénotype cardiaque hérité par la lignée maternelle et la transmission des variantes CAPN3 et LMNA à travers les membres de la famille.
Au total, ces données soulèvent des considérations importantes. Tout d’abord, le panel NGS chez ce patient était essentiel pour atteindre le diagnostic moléculaire de la calpainopathie, en détectant une mutation connue et un deuxième nouveau variant prédit comme pathogène. Deuxièmement, le panneau NGS a permis l’identification de la nouvelle variante LMNA_c.550C > T dans le proband. Ce résultat était cohérent avec les antécédents familiaux positifs et avec les données de ségrégation, expliquant les maladies cardiovasculaires chez la mère et, plus important encore, recommandant un suivi cardiologique plus spécifique dans le proband. Il est important de noter que les manifestations cardiologiques se sont produites chez la mère plus tard dans l’âge (55 ans), de sorte que nous pouvons supposer que le proband pourrait encore être dans une « phase cardiologique asymptomatique. »Cependant, une expression phénotypique variable de la mutation LMNA dans le proband par rapport à la mère ne peut être exclue. Toutes ces données soulignent l’importance d’une approche intégrée entre cliniciens et généticiens, pour une interprétation correcte des résultats, un conseil génétique approprié et, à terme, une prise en charge et un suivi cliniques. En ce qui concerne le conseil génétique et familial, l’analyse NGS a également été réalisée sur le partenaire du proband, afin d’estimer le risque reproductif pour le couple. Le partenaire était négatif pour l’ensemble des 18 gènes testés, ce qui signifie qu’il a un risque résiduel de 1/650 d’être un porteur sain de mutations causales de la LGMD, étant donné que le test est sensible à 84%. Compte tenu du profil génétique du proband et de la sensibilité du test NGS, le risque résiduel pour le couple d’avoir un enfant atteint de calpainopathie est de 1/1300. D’autre part, les variants pathogènes LMNA sont transmis selon un schéma autosomique dominant, ce qui signifie que le proband a 50% de probabilité d’avoir des enfants hétérozygotes. Cependant, le tableau clinique de la progéniture ne peut être certainement prédit car le LMNA_c.550C > T est une nouvelle variante et son impact fonctionnel sur le phénotype est encore inconnu.
En conclusion, ce rapport de cas met en évidence l’utilité clinique des panels NGS pour fournir un diagnostic précis de la LGMD2A et décrire des phénotypes et des comorbidités complexes provenant de l’héritage de différentes mutations dans plusieurs gènes. Cependant, l’application de NGS dans la pratique clinique doit toujours être associée à un conseil pré- et post-génétique afin de fournir une explication claire des résultats, des implications possibles sur le phénotype des patients, le risque de récidive au sein de la famille ainsi que d’expliquer d’éventuels résultats inattendus.
Disponibilité des données
Aucun ensemble de données n’a été généré ou analysé pour cette étude.
Déclaration d’éthique
Cette étude a été réalisée conformément aux recommandations du comité d’éthique de la Fondation Santa Lucia avec le consentement éclairé écrit de tous les sujets. Tous les sujets ont donné leur consentement écrit en connaissance de cause conformément à la Déclaration d’Helsinki. Le protocole a été approuvé par le comité d’éthique de la Fondation Santa Lucia.
Contributions des auteurs
RC, CSt, VC, GC, RG, GPa, SC, CP et JM ont contribué à l’acquisition de données, à l’analyse et à l’interprétation des données. CSt, RC, GC, RG, GM et EG ont participé à la rédaction du manuscrit. SZ, GPr, CSa et SS ont participé à l’acquisition de données cliniques. RC, CSt, VC, GC, RG, GPa, SC, CP, JM, SZ, GPr, GM, CSa, SS et EG ont donné l’approbation finale de la version à publier.
Financement
Ce travail est soutenu par 5X 2016 Ministère national de la Santé 2.
Déclaration sur les conflits d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de relations commerciales ou financières pouvant être interprétées comme un conflit d’intérêts potentiel.
Matériel supplémentaire
Le matériel supplémentaire pour cet article peut être trouvé en ligne à l’adresse suivante:: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. Diagnostic protéique et génétique de la dystrophie musculaire de la ceinture des membres de type 2A: le rendement et les pièges. Nerf musculaire. (2015) 52:163–73. doi: 10.1002/mus.24682
Résumé PubMed / Texte intégral CrossRef / Google Scholar
2. Nigro V, Savarese M. Base génétique des dystrophies musculaires de la ceinture des membres: la mise à jour de 2014. Acta Myol. (2014) 33:1–12.
Résumé PubMed / Google Scholar
3. Richard I, Hogrel JY, Stockholm D, Payan CAM, Fougerousse F, Eymard B, et al. Natural history of LGMD2A for delinating outcome measures in clinical trials. Ann Clin Transl Neurol. (2016) 3:248–65. doi: 10.1002/acn3.287
Résumé PubMed / Texte intégral CrossRef / Google Scholar
4. Angelini C, Fanin M. Calpainopathie. Dans: Adam MP, Ardinger HH, éditeurs Pagon RA. GeneReviews®. Seattle, WA: Université de Washington (2017) 1-30.
Résumé PubMed / Google Scholar
5. Kyriakides T, Angelini C, Schaefer J, Sacconi S, Siciliano G, Vilchez JJ, et al. Lignes directrices EFNS sur l’approche diagnostique de l’hyperckémie pauci- ou asymptomatique. Eur J Neurol. (2010) 17:767–73. doi: 10.1111/j.1468-1331.2010.03012.x
CrossRef Texte intégral / Google Scholar
6. Mori-Yoshimura M, Segawa K, Minami N, Oya Y, Komaki H, Nonaka I, et al. Dysfonctionnement cardiopulmonaire chez les patients atteints de dystrophie musculaire de la ceinture des membres 2A. Nerf musculaire. (2017) 55:465–9. doi: 10.1002/mus.25369
CrossRef Texte intégral / Google Scholar
7. Okere A, Reddy SS, Gupta S, Shinnar M. Une cardiomyopathie chez un patient atteint de dystrophie musculaire de la ceinture des membres de type 2A. Insuffisance cardiaque Circ. (2013) 6:e12–13. doi: 10.1161/ CIRCHEARTFAILURE.112.971424
CrossRef Texte intégral / Google Scholar
8. Kramerova I, Ermolova N, Eskin A, Hevener A, Quehenberger O, Armando AM, et al. L’incapacité à réguler la transcription des gènes nécessaires à l’adaptation musculaire sous-tend la dystrophie musculaire de la ceinture des membres 2A (calpainopathie). Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093/hmg/ddw086
Résumé PubMed / Texte intégral Croisé / Google Scholar
9. Thompson R, Straub V. Dystrophies musculaires de la ceinture des membres – collaborations internationales pour la recherche translationnelle. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038/nrneurol.2016.35
Résumé PubMed / Texte intégral CrossRef / Google Scholar
10. Strafella C, Caputo V, Galota MR, Zampatti S, Marella G, Mauriello S, et al. Application de la médecine de précision dans les maladies neurodégénératives. Neurol avant. (2018) 9:701. doi: 10.3389 / fneur.2018.00701
Résumé PubMed / Texte intégral CrossRef / Google Scholar
11. Cascella R, Strafella C, Caputo V, Errichiello V, Zampatti S, Milano F, et al. Vers l’application de la médecine de précision dans la dégénérescence maculaire liée à l’âge. Prog Retin Eye Res. (2017) 63:132-46. doi: 10.1016 / j. preteyeres.2017.11.004
Résumé PubMed / Texte intégral CrossRef / Google Scholar
12. Cascella R, Strafella C, Longo G, Manzo L, Ragazzo M, De Felici C, et al. Évaluer le risque individuel de DMLA avec des conseils génétiques, des antécédents familiaux et des tests génétiques. Oeil. (2017) 32:446–50. doi: 10.1038/ oeil.2017.192
Résumé PubMed / Texte intégral CrossRef / Google Scholar
13. Adams DR, Eng CM. Séquençage de nouvelle génération pour diagnostiquer des troubles génétiques présumés. En anglais J Med. (2019) 379:1353–62. doi: 10.1056 / NEJMc1814955
Résumé publié / Texte intégral croisé / Google Scholar
14. Angelini C. Maladie neuromusculaire. Diagnostic et découverte de la dystrophie musculaire de la ceinture des membres. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038/nrneurol.2015.230
CrossRef Texte intégral / Google Scholar
15. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, et al. Utilisation du séquençage de l’exome entier pour le diagnostic de la dystrophie musculaire de la ceinture des membres: résultats et leçons apprises. JAMA Neurol. (2015) 72:1424–32. doi: 10.1001 / jamaneurol.2015.2274
Résumé PubMed / Texte intégral CrossRef / Google Scholar
16. Milic A, Daniele N, Lochmüller H, Mora M, Comi GP, Moggio M, et al. Un tiers des biopsies de LGMD2A ont une activité protéolytique normale de la calpaïne 3, telle que déterminée par un test in vitro. Troubles Neuromusques. (2007) 17:148–56. doi: 10.1016/j.nmd.2006.11.001
Résumé PubMed / Texte intégral CrossRef / Google Scholar
17. Lanzillo R, Aurino S, Fanin M, Aguennoz M, Vitale F, Fiorillo C, et al. Calpainopathie à début précoce avec un niveau normal de calpain 3 non fonctionnel. Dev Med Neurol Enfant. (2006) 48:304–6. doi: 10.1017/S001216220600065X
Résumé PubMed / Texte intégral Croisé / Google Scholar
18. Talim B, Ognibene A, Mattioli E, Richard I, Anderson LV, Merlini L. Expression normale de la calpaïne dans la dystrophie musculaire de la ceinture des membres génétiquement confirmée de type 2A. Neurologie. (2001) 56:692–3. doi: 10.1212/wnl.56.5.692-a
CrossRef Texte intégral / Google Scholar
19. Díaz-Manera J, Llauger J, Gallardo E, Illa I. IRM musculaire dans les dystrophies musculaires. Acta Myol. (2015) 34:95–108.
Résumé PubMed / Google Scholar
20. Mercuri E, Bushby K, Ricci E, Birchall DR, Pane M, Kinali M, et al. Résultats de l’IRM musculaire chez les patients atteints de dystrophie musculaire de la ceinture des membres avec déficit en calpaïne 3 (LGMD2A) et contractures précoces. Troubles Neuromusques. (2005) 15:164–71. doi: 10.1016/j.nmd.2004.10.008
Résumé PubMed / Texte intégral CrossRef / Google Scholar
21. Magri F, Nigro V, Angelini C, Mongini T, Mora M, Moroni I, et al. Registre italien de la dystrophie musculaire de la ceinture des membres: fréquence relative, caractéristiques cliniques et diagnostic différentiel. Nerf musculaire. (2017) 55:55–68. doi: 10.1002/mus.25192
Résumé PubMed / Texte intégral CrossRef / Google Scholar
22. Chae J, Minami N, Jin Y, Nakagawa M, Murayama K, Igarashi F, et al. Mutations du gène Calpain 3: résultats génétiques et clinico-pathologiques dans la dystrophie musculaire de la ceinture des membres. Troubles Neuromusques. (2001) 11:547–55. doi: 10.1016/S0960-8966(01)00197-3
Résumé PubMed / Texte intégral CrossRef / Google Scholar
23. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/ gim.2015.30
Résumé PubMed / Texte intégral CrossRef / Google Scholar
24. Bonne G, Mercuri E, Muchir A, Urtizberea A, Bécane HM, Recan D, et al. Spectre génétique clinique et moléculaire de la dystrophie musculaire autosomique dominante d’Emery-Dreifuss due à des mutations du gène lamin A/ C. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170:: AID-ANA6 >3.0.CO ;2-J
Résumé PubMed / Texte intégral CrossRef / Google Scholar
25. Hong JS, Ki CS, Kim JW, Suh YL, Kim JS, Baek KK, et al. Dysrythmies cardiaques, cardiomyopathie et dystrophie musculaire chez les patients atteints de dystrophie musculaire d’Emery-Dreifuss et de dystrophie musculaire de la ceinture des membres de type 1B.J Korean Med Sci. (2005) 20:283–90. doi: 10.3346/ jkms.2005.20.2.283
CrossRef Texte intégral / Google Scholar