- Introduction
- Méthodes
- Filtrage des données de profilage Ribosomique
- Assemblage de Génomes virtuels
- Détermination de la Région Lue-Mappée Ribo-seq sur une Jonction (RMRJ) de CIRCRNA
- Formation du Modèle et de la classification des RMRJ
- Prédiction des peptides traduits par les RMRJ
- Résultats et discussion
- circRNAs traduits chez l’Homme et A. thaliana
- Enrichissement fonctionnel des circRNAs Humains et A. thaliana Avec un Potentiel de codage
- Test de précision du CircCode
- Influence de la profondeur de séquençage des données Ribo-seq
- Comparaison du CircCode Avec d’Autres Outils
- Conclusions
- Disponibilité et exigences
- Déclaration de disponibilité des données
- Contributions de l’auteur
- Financement
- Conflit d’intérêts
- Matériel supplémentaire
Introduction
Les ARN circulaires (CIRCRNA) sont un type particulier de molécule d’ARN non codant qui est devenu un sujet de recherche brûlant dans le domaine de l’ARN et qui reçoit beaucoup d’attention (Chen et Yang, 2015). Par rapport aux ARN linéaires traditionnels (contenant des extrémités 5′ et 3′), les molécules de circRNA ont généralement une structure circulaire fermée; les rendant plus stables et moins sujettes à la dégradation (Vicens et Westhof, 2014). Bien que l’existence de CIRCRNA soit connue depuis un certain temps, ces molécules ont été considérées comme un sous-produit de l’épissage de l’ARN. Cependant, avec le développement des technologies de séquençage et de bioinformatique à haut débit, les CIRCNAS sont devenus largement reconnus chez les animaux et les plantes (Chen et Yang, 2015). Des études récentes ont également montré qu’un grand nombre de circRNAs peuvent être traduits en petits peptides dans les cellules (Pamudurti et al., 2017) et ont des rôles clés malgré leur niveau d’expression parfois faible (Hsu et Benfey, 2018; Yang et al., 2018). Bien qu’un nombre croissant de circRNAs soient identifiés, leurs fonctions chez les plantes et les animaux restent généralement à étudier. En plus de leurs fonctions de leurres miARN, les CIRCRNA ont un potentiel de traduction important, mais aucun outil n’est disponible pour prédire spécifiquement les capacités de traduction de ces molécules (Jakobi et Dieterich, 2019).
Plusieurs outils existent pour la prédiction et l’identification des CIRCNAS, tels que CIRI (Gao et al., 2015), CIRCexplorer (Dong et al., 2019), CircPro (Meng et coll., 2017), et circtools (Jakobi et al., 2018). Parmi eux, CircPro peut révéler des circRNAs traduits en calculant un score de potentiel de traduction pour les circRNAs basé sur CPC (Kong et al., 2007), qui est un outil permettant d’identifier le cadre de lecture ouvert (ORF) dans une séquence donnée. Cependant, parce que certains CIRCRNA n’utilisent pas le codon de départ lors de la traduction (Ingolia et al., 2011; Slavoff et coll., 2013; Kearse et Wilusz, 2017; Spealman et coll., 2018), l’utilisation de CPC peut filtrer certains circRNAs vraiment traduits. Dans cette étude, nous avons utilisé BASiNET (Ito et al., 2018), qui est un classificateur d’ARN basé sur les méthodes d’apprentissage automatique (forêt aléatoire et modèle J48). Il transforme d’abord les ARN codants (données positives) et les ARN non codants (données négatives) donnés et les représente comme des réseaux complexes; il extrait ensuite les mesures topologiques de ces réseaux et construit un vecteur caractéristique pour entraîner le modèle utilisé pour classer la capacité de codage des CIRCRNA. Avec cette méthode, le filtrage erroné des circRNAs traduits qui ne sont pas initiés par AUG est évité. De plus, la technologie Ribo-seq, basée sur le séquençage à haut débit pour surveiller les RPF (fragments protégés ribosomaux) des transcriptions (Guttman et al., 2013; Brar et Weissman, 2015), peuvent être utilisés pour déterminer les emplacements des circRNAs en cours de traduction (Michel et Baranov, 2013). Pour identifier la capacité de codage des circRNAs, nous avons développé l’outil CircCode, qui implique un framework basé sur Python 3, et appliqué CircCode pour étudier le potentiel de traduction des circRNAs des humains et d’Arabidopsis thaliana. Nos travaux fournissent une riche ressource pour une étude plus approfondie des fonctions des CIRCRNA avec une capacité de codage.
Méthodes
CircCode a été écrit dans le langage de programmation Python 3 ; il utilise Trimmomatic (Bolger et al., 2014), bowtie (Langmead et Salzberg, 2012) et STAR (Dobin et al., 2013) pour filtrer les lectures brutes de Ribo-seq et mapper ces lectures filtrées au génome. Le CircCode identifie ensuite les régions en lecture mappées Ribo-seq dans les CIRCRNA qui contiennent des jonctions. Après cela, les séquences candidates mappées dans les CIRCNAS sont triées en fonction des classificateurs (modèle J48) en ARN codants et en ARN non codants par BASiNET. Enfin, les peptides courts produits par traduction sont identifiés comme des régions codantes potentielles des CIRCRNA. L’ensemble du processus de CircCode se compose de cinq étapes (Figure 1).
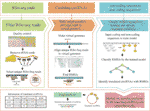
Figure 1 Le flux de travail de CircCode. La couche supérieure représente le fichier d’entrée requis pour chaque étape du CircCode. La couche intermédiaire est divisée en trois parties et chaque partie représente une étape de fonctionnement différente. De gauche à droite, la première partie représente le filtrage des données Ribo-seq ; le contrôle qualité est exécuté par Trimmomatic, et les lectures d’ARNr sont supprimées par bowtie. La deuxième partie représente les étapes utilisées pour produire le génome virtuel et aligner les lectures filtrées sur le génome virtuel avec STAR. La dernière partie représente l’identification des circRNAs traduits par apprentissage automatique. La couche inférieure représente la dernière étape utilisée pour prédire les peptides traduits à partir des CIRCRNA et les résultats finaux de sortie, y compris les informations sur les CIRCRNA traduits et leurs produits de traduction.
Filtrage des données de profilage Ribosomique
Tout d’abord, les fragments et adaptateurs de mauvaise qualité dans les lectures Ribo-Seq sont supprimés par Trimmomatic avec les paramètres par défaut pour obtenir des lectures Ribo-seq propres. Deuxièmement, ces lectures Ribo-seq propres sont mappées à une bibliothèque d’ARNr pour supprimer les lectures dérivées de l’ARNr à l’aide de bowtie. Les longueurs de lecture de Ribo-seq étant relativement courtes (généralement inférieures à 50 pb), il est possible qu’une lecture corresponde à plusieurs régions. Dans ce cas, il est difficile de déterminer à quelle région correspond une lecture particulière. Pour éviter cela, les lectures Ribo-seq propres sont mappées au génome d’une espèce d’intérêt, et les lectures qui ne sont pas parfaitement alignées sur le génome sont considérées comme les lectures Ribo-seq uniques finales.
Assemblage de Génomes virtuels
Les CircRNAs apparaissent généralement sous forme de molécules en forme d’anneau chez les eucaryotes, et ils peuvent être identifiés en fonction de leurs jonctions d’épissage arrière. Cependant, les séquences de circRNAs dans le fichier fasta sont souvent sous forme linéaire. En théorie, le résultat indique que la jonction est entre le nucléotide terminal 5′ et le nucléotide terminal 3′, bien que la jonction et la séquence près de la jonction ne puissent pas être vues directement, alignant ainsi les lectures Ribo-seq sur les séquences circNNA, y compris les jonctions, de manière simple.
Le CircCode relie la séquence de chaque CIRCNA en tandem de sorte que la jonction de chacun se trouve au milieu de la séquence nouvellement construite. Nous avons également séparé chaque unité de série de 100 N nucléotides pour éviter toute confusion à l’étape d’alignement des séquences (la longueur de chaque FPR est inférieure à 50 pb). Enfin, nous avons obtenu un génome virtuel constitué uniquement de CIRCRNA candidats en tandem séparés de 100 Ns. Étant donné que le CircCode se concentre uniquement sur l’alignement entre les lectures Ribo-seq et les séquences de circRNA, nous pouvons étudier le potentiel de codage des CIRCRNA en mappant les lectures Ribo-seq sur ce génome virtuel, ce qui peut économiser beaucoup de temps de calcul (le génome virtuel est beaucoup plus petit que le génome entier) et augmenter la précision (en évitant les interférences entre les comparaisons de séquences amont et aval des CIRCRNA).
Détermination de la Région Lue-Mappée Ribo-seq sur une Jonction (RMRJ) de CIRCRNA
Les lectures uniques finales de Ribo-seq sont mappées sur un génome virtuel créé précédemment à l’aide d’ÉTOILES. Étant donné que chaque unité circRNA tandem était séparée par 100 N bases avant de produire le génome virtuel, la plus grande longueur d’intron a été réglée pour ne pas dépasser 10 bases avec le paramètre « –alignIntronMax 10. »Ce paramètre élimine toute interaction entre différents circRNAs dans l’alignement de la séquence. Dans la deuxième étape de production de génome virtuel, le CircCode stocke des informations de jonction positionnelle pour chaque circRNA dans le génome virtuel. Si la région cartographiée en lecture Ribo-seq dans le génome virtuel inclut la jonction du circRNA, et que le nombre de lectures Ribo-seq cartographiées à la jonction (NMJ) est supérieur à 3, la région cartographiée en lecture Ribo-seq à la jonction des CIRCRNA peut être considérée comme un RMRJ, qui révèle un segment grossièrement traduit des CIRCRNA près du site de jonction.
Formation du Modèle et de la classification des RMRJ
Bien que les RMRJ puissent constituer une preuve puissante de la traduction, cette méthode présente encore quelques lacunes. Comme la longueur des lectures de la carte ribosomique est courte, une lecture peut être comparée à la mauvaise position. Par conséquent, il n’est pas convaincant de considérer simplement la région couverte par le Ribo-seq comme la région traduite. À cette fin, la méthode d’apprentissage automatique est utilisée pour identifier la capacité de codage du RMRJ. Tout d’abord, CircCode extrait les ARN codants (données positives) et les ARN non codants (données négatives) d’une espèce d’intérêt et les utilise pour l’apprentissage du modèle au moyen de la différence de vecteurs caractéristiques entre les ARN codants et les ARN non codants. CircCode utilise ensuite le modèle entraîné pour classer les RMRJ obtenus à l’étape précédente par BASiNET. Si le RMRJ d’un circRNA est reconnu comme ARN codant, alors ce circRNA peut être identifié comme un circRNA traduit.
Prédiction des peptides traduits par les RMRJ
Comme l’expression des CIRCRNA dans les organismes est faible, les données Ribo-seq ne montrent pas clairement la périodicité exacte de 3 nt dans le cas de moins de RPF. Par conséquent, il est difficile de déterminer le site de début de traduction exact d’un circRNA traduit. En raison de la présence d’un codon d’arrêt dans certains RMRJ et parce que le codon de départ est difficile à déterminer, la méthode de recherche d’un ORF basé sur un codon de départ et un codon d’arrêt n’est pas réalisable.
Pour déterminer les véritables régions de traduction de ces circRNAs et générer le produit de traduction final, FragGeneScan (Rho et al., 2010), qui peut prédire des régions codant des protéines dans des gènes fragmentés et des gènes avec des décalages de cadre, est utilisé pour déterminer les peptides traduits produits par les CIRCRNA.
Pour éviter le processus d’exécution fastidieux, tous les modèles peuvent être appelés par un script shell; l’utilisateur peut simplement remplir le fichier de configuration donné et l’entrer dans le script, et l’ensemble du processus de prédiction des circRNAs traduits sera alors exécuté. De plus, CircCode peut être exécuté séparément, étape par étape, de sorte que l’utilisateur puisse ajuster les paramètres au milieu de la procédure et afficher les résultats de chaque étape comme souhaité.
Résultats et discussion
Après des tests sur plusieurs ordinateurs, CircCode s’est avéré fonctionner correctement avec les dépendances requises installées. Pour tester les performances de CircCode, nous avons utilisé des données pour les humains et A. thaliana pour prédire les circRNAs avec un potentiel de traduction. Les résultats ont été comparés avec des CIRCNAS qui ont été vérifiés expérimentalement comme confirmation. Par la suite, nous avons testé la valeur du taux de fausse découverte (FDR) de CircCode. Nous avons utilisé des GenRGenS (Ponty et al., 2006) pour générer un ensemble de données à tester sur la base de circRNAs traduits connus et confirmer que la valeur du FDR se situait dans une plage acceptable et à un niveau faible. Enfin, nous avons évalué l’effet de différentes profondeurs de séquençage des données Ribo-seq sur les prédictions de CircCode et comparé CircCode à d’autres logiciels.
circRNAs traduits chez l’Homme et A. thaliana
Pour appliquer l’outil CircCode à des données réelles, nous avons d’abord téléchargé les fichiers comprenant le génome de référence humain GRCh38, l’annotation du génome et l’ARNr humain, depuis Ensembl. Pour A. thaliana, les génomes de référence (TAIR10), les fichiers d’annotation du génome et les séquences d’ARNr correspondantes ont tous été téléchargés à partir de plantes Ensembl. Les données Ribo-seq pour les humains et A. thaliana ont été téléchargées à partir de la RPFdb (numéros d’accession : GSE96643, GSE81295, GSE88794) (Hsu et al., 2016; Willems et coll., 2017), et tous les circRNAs candidats de human et A. thaliana ont été téléchargés à partir de CIRCPedia v2 (Dong et al., 2018) et Plantcirbase, respectivement (Chu et al., 2017). En fin de compte, nous avons identifié 3 610 circRNAs traduits d’humains et 1 569 circRNAs traduits d’A. thaliana à l’aide du CircCode (Données supplémentaires 1).
Enrichissement fonctionnel des circRNAs Humains et A. thaliana Avec un Potentiel de codage
En utilisant les résultats du CircCode pour l’homme et A. thaliana, l’outil en ligne KOBAS 3.0 (Wu et al., 2006) a été utilisé pour annoter ces circRNAs traduits en fonction de leurs gènes parents. De plus, nous avons effectué une analyse fonctionnelle GO (Ontologie des gènes) et une analyse d’enrichissement KEGG (Encyclopédie de Kyoto des gènes et des génomes) pour ces CIRCNAS traduits à l’aide du clusterProfiler de paquets R (Yu et al., 2012).
Les résultats de KEGG ont montré que les CIRCRNA humains étaient enrichis en traitement protéique dans la voie du réticulum endoplasmique, la voie du métabolisme du carbone et la voie du transport de l’ARN. L’analyse GO a indiqué la participation des CIRCRNA traduits chez l’homme à la régulation de la liaison aux molécules, de l’activité des ATPASES et d’autres processus biologiques liés à l’épissage de l’ARN. De plus, les circRNAs traduits d’A. thaliana sont enrichis en voies liées à la résistance au stress, suggérant qu’ils jouent un rôle essentiel dans ce processus (Données supplémentaires 2).
Test de précision du CircCode
Pour étudier la précision du CircCode, des séquences de test générées par GenRGenS, qui utilise le modèle de Markov caché pour produire des séquences ayant les mêmes caractéristiques de séquence (telles que les fréquences de différents nucléotides, différents codons et différents nucléotides au début de la séquence), ont été utilisées.
Pour cette étude, nous avons utilisé des circRNAs traduits chez l’homme précédemment publiés (Yang et al., 2017) comme entrée pour GenRGenS et généré 10 000 séquences pour tester le CircCode. Nous avons répété le test 10 fois, et en moyenne, 27 circRNAs traduits ont été prédits à chaque fois. La valeur du FDR a été calculée à 0,0027, ce qui est beaucoup moins que 0,05, ce qui indique que les résultats prévus sont crédibles.
De plus, nous avons comparé les CIRCRNA traduits d’humains identifiés par le CircCode avec des données vérifiées sur les circRNA associés aux polysomes (Yang et al., 2017). Parmi eux, 60% des CIRCRNA ont été identifiés par CircCode (Données supplémentaires 3).
Influence de la profondeur de séquençage des données Ribo-seq
Pour étudier l’impact de la profondeur de séquençage des données Ribo-seq sur les résultats d’identification du CircCode, nous avons d’abord testé l’effet de la profondeur de séquençage sur le nombre de circRNAs traduits (Figure 2A). Lorsque la profondeur de séquençage était faible, le nombre prédit de circRNAs traduits était faible et le nombre de circRNAs traduits augmentait avec l’augmentation de la profondeur de séquençage. Le nombre de circRNAs traduits est devenu stable lorsque la profondeur de séquençage a atteint pas moins de 10× couverture de transcription linéaire.

Figure 2 (A) Effet de la profondeur de séquençage des données Ribo-seq sur le nombre prédit de circRNAs traduits. (B) L’effet du nombre de lecture de jonction (JRN) sur la sensibilité du CircCode à différentes profondeurs de séquençage.
Deuxièmement, l’influence de la NMJ sur la sensibilité à différentes profondeurs de séquençage a également été évaluée (figure 2B). Les résultats ont montré que la NMJ avait moins d’impact sur la sensibilité à mesure que la profondeur de séquençage augmentait. CircCode avait également une sensibilité plus élevée lors de l’utilisation de données Ribo-seq avec une profondeur de séquençage plus élevée.
Comparaison du CircCode Avec d’Autres Outils
Pour comparer le CircCode avec d’autres outils, tels que CircPro, le même ensemble de données Ribo-seq (SRR3495999) d’A. thaliana a été utilisé pour identifier les CIRCRNA traduits à l’aide de six processeurs, avec 16 gigaoctets de RAM. CircPro a identifié 44 circRNAs traduits en 13 min, tandis que CircCode en a identifié 76 en 20 min. Ainsi, CircCode est plus sensible que CircPro au même niveau de matériel informatique, mais cela prend plus de temps. CircPro est concis et prend moins de temps que CircCode, mais CircCode peut identifier plus de CIRCRNA avec une capacité de codage que CircPro.
Conclusions
Les CircRNAs jouent un rôle important en biologie, et il est crucial d’identifier avec précision les circRNAs ayant une capacité de codage pour des recherches ultérieures. Sur la base de Python 3, nous avons développé CircCode, un outil de ligne de commande facile à utiliser qui a une grande sensibilité pour identifier les circRNAs traduits à partir de lectures Ribo-Seq avec une grande précision. CircCode présente de bonnes performances chez les plantes et les animaux. Les travaux futurs ajouteront l’analyse des caractères en aval au CircCode en visualisant chaque étape du processus et en optimisant la précision de la prédiction.
Disponibilité et exigences
CircCode est disponible à https://github.com/PSSUN/CircCode; système(s) d’exploitation: Linux, langages de programmation: Python 3 et R; autres exigences: bedtools (version 2.20.0 ou ultérieure), bowtie, STAR, paquets Python 3 (Biopython, Pandas, rpy2), paquets R (BASiNET, Biostrings). Les packages d’installation de tous les logiciels requis sont disponibles sur la page d’accueil de CircCode. Les utilisateurs n’ont pas besoin de les télécharger individuellement. La page d’accueil de CircCode fournit également des manuels d’utilisation détaillés pour référence. L’outil est disponible gratuitement. Il n’y a pas de restrictions sur l’utilisation par les nonacadémiques.
Déclaration de disponibilité des données
Toutes les données pertinentes se trouvent dans le manuscrit et les fichiers d’information à l’appui.
Contributions de l’auteur
Conceptualisation: PS, GL. Conservation des données: PS, GL. Analyse formelle: PS, GL. Rédaction – Brouillon original: PS, GL. Rédaction – Révision et édition: PS, GL.
Financement
Ce travail a été soutenu par des subventions de la Fondation Nationale des Sciences Naturelles de Chine (subventions nos 31770333, 31370329 et 11631012), du Programme pour les Excellents Talents du Nouveau Siècle à l’Université (NCET-12-0896) et des Fonds de Recherche Fondamentale pour les Universités Centrales (no. GK201403004). Les organismes de financement n’ont joué aucun rôle dans l’étude, sa conception, la collecte et l’analyse des données, la décision de publier ou la préparation du manuscrit. Les bailleurs de fonds n’ont joué aucun rôle dans la conception de l’étude, la collecte et l’analyse des données, la décision de publier ou la préparation du manuscrit.
Conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de relations commerciales ou financières pouvant être interprétées comme un conflit d’intérêts potentiel.
Matériel supplémentaire
Le matériel supplémentaire pour cet article peut être trouvé en ligne à l’adresse suivante:: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
Données supplémentaires 1/ La séquence du circRNA traduit prédit et du peptide court.
Données supplémentaires 2/ Résultats de l’enrichissement en GO et de l’enrichissement en FÛTS pour l’homme et Arabidopsis thaliana.
Données supplémentaires 3/ Comparaison des circRNAs traduits prédits avec des circRNAs traduits validés.
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: un trimmer flexible pour les données de séquence illumina. Bioinformatique 30, 2114-2120. doi: 10.1093 / bioinformatique / btu170
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Il s’agit de la première édition de la série. Le profilage des ribosomes révèle le quoi, quand, où et comment de la synthèse des protéines. NAT. Rév. Mol. Biol cellulaire. 16, 651–664. doi: 10.1038 / nrm4069
Résumé PubMed / Texte intégral Croisé / Google Scholar
Chen, L.-L., Yang, L. (2015). Régulation de la biogenèse du circRNA. Biol d’ARN. 12, 381–388. doi: 10.1080/15476286.2015.1020271
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Chu, Q., Zhang, X., Zhu, X., Liu, C., Mao, L., Ye, C., et al. (2017). Plantcirbase: une base de données pour les ARN circulaires des plantes. Mol. Usine 10, 1126-1128. doi: 10.1016/j.molp.2017.03.003
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jai, S., et al. (2013). ÉTOILE : aligneur universel ultra-rapide d’ARN-seq. Bioinformatique 29, 15-21. doi: 10.1093 / bioinformatique / bts635
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Dong, R., Ma, X.-K., Chen, L.-L., Yang, L. (2019). « Annotation à l’échelle du génome des CIRCRNA et de leur épissage / épissage alternatif avec pipeline CIRCexplorer », dans Eitranscriptomics. EDS. Wajapeyee, N., Gupta, R. (New York, NY: Springer New York), 137-149. doi: 10.1007/978-1-4939-8808-2_10
CrossRef Texte intégral / Google Scholar
Dong, R., Ma, X.-K., Li, G.-W., Yang, L. (2018). CIRCpedia v2: une base de données mise à jour pour l’annotation circulaire complète de l’ARN et la comparaison des expressions. Génomique Protéomique Bioinf. 16, 226–233. doi: 10.1016/j.gpb.2018.08.001
CrossRef Texte intégral / Google Scholar
Gao, Y., Wang, J., Zhao, F. (2015). CIRI: un algorithme efficace et impartial pour l’identification d’ARN circulaire de novo. Biol génomique. 16, 4. doi: 10.1186/s13059-014-0571-3
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Guttman, M., Russell, P., Ingolia, N. T., Weissman, J. S., Lander, E. S. (2013). Le profilage des ribosomes fournit la preuve que les ARN non codants de grande taille ne codent pas pour les protéines. Cellule 154, 240-251. doi: 10.1016/j. cell.2013.06.009
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Hsu, P.Y., Benfey, P.N. (2018). Petit mais puissant: peptides fonctionnels codés par de petits ORF dans les plantes. PROTÉOMIQUE 18, 1700038. doi: 10.1002/ pmic.201700038
CrossRef Texte intégral / Google Scholar
Hsu, P. Y., Calviello, L., Wu, H.-Y. L., Li, F.-W., Rothfels, C. J., Ohler, U., et al. (2016). Le profilage des ribosomes à super résolution révèle des événements de traduction non notés dans Arabidopsis. Proc. Natl. Acad. Sci. 113, E7126–E7135. doi: 10.1073/pnas.1614788113
CrossRef Texte intégral / Google Scholar
Ingolia, N.T., Lareau, L. F., Weissman, J. S. (2011). Le profilage des ribosomes de cellules souches embryonnaires de souris révèle la complexité et la dynamique des protéomes de mammifères. Cellule 147, 789-802. doi: 10.1016/j. cell.2011.10.002
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Ito, E. A., Katahira, I., Vicente, F. F., da, R., Pereira, L. F. P., Lopes, F. M. (2018). BASiNET – Réseau de séquences biologiques : étude de cas sur l’identification des ARN codants et non codants. Acides nucléiques Res. 46, e96–e96. doi: 10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093/ bioinformatique / bty948
CrossRef Texte intégral / Google Scholar
Kearse, M. G., Wilusz, J. E. (2017). Traduction non-AUG: un nouveau départ pour la synthèse des protéines chez les eucaryotes. Gènes Dev. 31, 1717–1731. doi: 10.1101/gad.305250.117
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Kong, L., Zhang, Y., Ye, Z.-Q., Liu, X.-Q., Zhao, S.-Q., Wei, L., et al. (2007). CPC: évaluer le potentiel de codage des protéines des transcrits à l’aide de caractéristiques de séquence et d’une machine vectorielle de support. Acides nucléiques Res. 35, W345-W349. doi: 10.1093/nar/gkm391
Résumé PubMed / Texte intégral Croisé / Google Scholar
Langmead, B., Salzberg, S. L. (2012). Alignement de lecture rapide avec Bowtie 2. NAT. Méthodes 9, 357-359. doi: 10.1038/nmeth.1923
Résumé PubMed / CrossRef Texte intégral / Google Scholar
Meng, X., Chen, Q., Zhang, P., Chen, M. (2017). CircPro : un outil intégré pour l’identification des CIRCRNA à potentiel de codage des protéines. Bioinformatique 33, 3314-3316. doi: 10.1093 / bioinformatique / btx446
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Michel, A. M., Baranov, P. V. (2013). Profilage des ribosomes: un moniteur Hi-Def pour la synthèse des protéines à l’échelle du génome: profilage des ribosomes. Wiley Interdiscip. Rev. ARN 4, 473-490. doi: 10.1002/ wrna.1172
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Pamudurti, N. R., Bartok, O., Jens, M., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., et al. (2017). Traduction des CircRNAs. Mol. Cellule 66, 9-21.e7. doi: 10.1016 / j. molcel.2017.02.021
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Ponty, Y., Termier, M., Denise, A. (2006). GenRGenS : logiciel de génération de séquences et de structures génomiques aléatoires. Bioinformatique 22, 1534-1535. doi: 10.1093/ bioinformatique / btl113
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Rho, M., Tang, H., Ye, Y. (2010). FragGeneScan: prédire les gènes dans des lectures courtes et sujettes aux erreurs. Acides nucléiques Res. 38, e191–e191. doi: 10.1093/nar/gkq747
Résumé PubMed / Texte intégral Croisé / Google Scholar
Il s’agit de l’une des principales sources d’approvisionnement en eau potable de la région. (2013). Peptidomic discovery of short open reading frame-coded peptides in human cells. NAT. Chem. Biol. 9, 59–64. doi: 10.1038 / nchembio.1120
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Spealman, P., Naik, A. W., May, G. E., Kuersten, S., Freeberg, L., Murphy, R. F., et al. (2018). UORF non AUG conservées révélées par une nouvelle analyse de régression des données de profilage des ribosomes. Genome Res. 28, 214-222. doi: 10.1101/gr.221507.117
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Vicens, Q., Westhof, E. (2014). Biogenèse des ARN circulaires. Cellule 159, 13-14. doi: 10.1016/j. cell.2014.09.005
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Willems, P., Ndah, E., Jonckheere, V., Stael, S., Sticker, A., Martens, L., et al. (2017). La protéomique N-terminale a aidé au profilage du paysage d’initiation de la traduction inexploré chez Arabidopsis thaliana. Mol. Cellule. Protéomique 16, 1064-1080. doi: 10.1074/ mcp.M116.066662
Résumé PubMed / Texte intégral Croisé / Google Scholar
Wu, J., Mao, X., Cai, T., Luo, J., Wei, L. (2006). Serveur KOBAS: une plate-forme Web pour l’annotation automatisée et l’identification des voies. Acides nucléiques Res. 34, W720-W724. doi: 10.1093/nar/gkl167
Résumé publié / Texte intégral croisé / Google Scholar
Yang, L., Fu, J., Zhou, Y. (2018). Les ARN Circulaires et leurs Rôles Émergents dans la Régulation immunitaire. Devant. Immunol. 9, 2977. doi: 10.3389 / fimmu.2018.02977
Résumé PubMed / Texte intégral CrossRef / Google Scholar
Yang, Y., Fan, X., Mao, M., Song, X., Wu, P., Zhang, Y., et al. (2017). Traduction étendue des ARN circulaires entraînés par la N6-méthyladénosine. Cellule 27, 626-641. doi: 10.1038/cr.2017.31
Résumé PubMed / Texte intégral CrossRef / Google Scholar
(2012). Clusterprofileur: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar