Importance
Les CIRCRNA suscitent un intérêt croissant en raison de leur implication dans de nombreux processus biologiques et maladies en plus de leur potentiel de biomarqueurs. Ils sont principalement détectés par la présence de lectures cartographiant leur jonction de rétrodiffusion. Néanmoins, les CIRCRNA ne sont plus les seuls transcrits contenant une telle jonction puisque des études récentes ont révélé que les ADN circulaires sont courants et peuvent être transcrits, ce qui donne des transcrits qui imiteraient un signal circRNA. Par conséquent, ce nouveau type de transcription chimérique peut changer la façon dont l’analyse du circNNA est effectuée et avoir un impact sur certains des résultats déjà rapportés.
Les ARN circulaires sont-ils les seuls Transcrits Chimériques ?
Les ARN circulaires (CIRCRNA) ont été redécouverts il y a quelques années sous forme de formes d’ARN non épissées canoniquement présentes dans différents organismes, y compris les humains (Salzman et al., 2012; Jeck et coll., 2013). Ce sont des transcrits fermés de manière covalente formés par un événement de rétroprojection d’ARN, où un donneur d’épissure d’un exon aval se joint à un accepteur d’épissure amont conduisant à des transcrits fermés de manière covalente qui sont caractérisés par la présence d’une jonction de rétroprojection qui distingue les CIRCRNA de leurs homologues linéaires (Figure 1A) (Zhang et al., 2016; Wilusz, 2018).
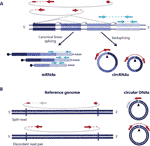
Figure 1 La détection de l’ARN circulaire et de l’ADN est basée sur des lectures couvrant la jonction. (A) La jonction rétrodiffusée entre les exons et la jonction canonique des exons sont représentées avec des lignes non continues et continues donnant respectivement lieu à des circRNAs et des ARNM. Les lectures d’extrémité appariées couvrant les jonctions en creux sont affichées en rouge et les lectures d’extrémité appariées compatibles avec une transcription linéaire sont affichées en bleu. (B) Comme les ARNCR, les ADN circulaires (ECCDNA) sont détectés sur la base de variantes de lecture structurelle compatibles avec un événement de circularisation représenté en rouge.
Depuis leur redécouverte, la communauté scientifique a attiré son attention sur les circRNAs et a étudié leur implication dans plusieurs processus cellulaires de santé et de maladie (Haque et Harries, 2017), leur rôle potentiel en tant que biomarqueurs (Abu et Jamal, 2016), et leurs fonctions de régulation (Floris et al., 2016). On sait maintenant que les CircRNAs sont abondants et stables dans le cytosol et le noyau (Salzman et al., 2012; Jeck et coll., 2013; Li et coll., 2015) et ont également été trouvés libres dans les biofluides (Bahn et al., 2015; Memczak et coll., 2015; Chen et coll., 2018) et dans les vésicules extracellulaires (Kyoung Mi et al., 2017). Le potentiel de biomarqueurs des circRNAs a été intensément étudié, en fait, de nombreuses études cas-témoins ont été publiées pour rechercher des circRNAs exprimés de manière différentielle qui pourraient être des biomarqueurs de différentes maladies. À ce jour, les circRNAs ont été impliqués dans plusieurs maladies, dont le cancer (Kristensen et al., 2017; Arnaiz et coll., 2018), troubles neurologiques (Akhter, 2018), maladies cardiovasculaires (Aufiero et al., 2019) et les maladies immunitaires (Iparraguirre et al., 2017; Liu et coll., 2019). Dans le même temps, comprendre pleinement leur biogenèse, leurs caractéristiques, leurs fonctions et leurs implications en biologie humaine reste une question ouverte pour les chercheurs dans le domaine.
Bien que la fonction de la plupart des circRNAs reste inconnue, il a été démontré que certains circRNAs peuvent agir comme des éponges de microARN, régulant les niveaux de microARN et leur activité (Hansen et al., 2013; Memczak et coll., 2013; Zheng et coll., 2016). Ils sont impliqués dans la régulation de l’expression génique en régulant la transcription de leurs gènes parentaux, en concurrence avec les protéines d’épissage linéaire ou d’épongage (Ashwal-Fluss et al., 2014; Li et coll., 2018). Fait intéressant, des études de profilage des ribosomes ont récemment montré que les circRNAs peuvent également être traduits in vitro et in vivo (Legnini et al., 2017; Yang et coll., 2017).
La caractéristique principale des circRNAs, et responsable de la plupart de leurs propriétés spéciales, est leur circularité. Par conséquent, en plus de détecter leur jonction rétroproject caractéristique, tester la circularité de ces molécules est l’un des points clés de chaque étude de circRNA. Néanmoins, de nombreuses études ont basé leur découverte des CIRCRNA sur l’ARN total et pourraient ainsi avoir interprété certains transcrits chimériques linéaires comme des CIRCRNA, entraînant des détections de circRNA faussement positives. Pour contourner ce problème, la plupart des études ont confirmé la circularité des transcrits trouvés par l’ARN-seq total en utilisant des méthodes RNase R, Northern blot ou électrophorétiques (Jeck et Sharpless, 2014). Cependant, ces validations de circularité ont aussi parfois révélé des transcriptions qui semblent être linéaires, plutôt que circulaires confirmant que la détection de CIRCARNAS à partir d’ARN total peut conduire à certains faux positifs. Ces faux positifs ont été attribués à des artefacts techniques ou à des transcriptions dérivées d’événements inhabituels tels que des duplications d’exons ou des événements de transplantation (Jeck et Sharpless, 2014; Szabo et Salzman, 2016). Cela dit, l’option d’avoir trouvé des transcrits linéaires vrais, biologiquement actifs et fonctionnels qui contiennent une séquence équivalente à une jonction rétrodiffusée (désormais appelés transcrits linéaires chimériques), a été quelque peu négligée car une source d’ARN linéaire de ce type n’était pas connue pour les cellules saines.
Les ADN circulaires comme source de transcriptions linéaires chimériques
La majeure partie du génome humain est organisée en chromosomes linéaires, cependant, certaines exceptions sont acceptées depuis longtemps, telles que l’ADN mitochondrial et les aberrations chromosomiques telles que les cercles d’ADN porteurs d’oncogènes (par exemple, les minutes doubles) (Benner et al., 1991; Nathanson et coll., 2014; Turner et coll., 2017) et les chromosomes annulaires (Tümer et al., 2004). Ce n’est que récemment que différents ADN circulaires tels que les micro-ADN (Shibata et al., 2012) ou des ADN circulaires extrachromosomiques (ECCDNA) ont également été trouvés provenant de grandes parties de différents génomes eucaryotes, y compris des humains et des levures (Møller et al., 2015; Kumar et coll., 2017; Møller et coll., 2018).
Les ADN circulaires sont formés lorsque deux extrémités d’un ADN linéaire sont jointes ensemble, ce qui donne une jonction similaire à la jonction rétrodiffusée sur les CIRCRNA communément appelée jonction de point d’arrêt qui est détectée sur la base de variantes de lecture structurelle compatibles avec un événement de circularisation (Gresham et al., 2010; Møller et coll., 2018; Prada-Luengo et al., 2019) (Figure 1B). Ils vont généralement d’une centaine de bases à des cercles de mégabases et peuvent contenir des exons et des gènes complets (Shibata et al., 2012; Møller et coll., 2015; Kumar et coll., 2017; Turner et coll., 2017; Møller et coll., 2018) et alors que certaines régions du génome se trouvent plus souvent sur l’ADN circulaire (Sinclair et Guarente, 1997;; Møller et al., 2016; Turner et coll., 2017; Møller et coll., 2018), la plupart des ADN circulaires semblent se produire au hasard (Shibata et al., 2012; Møller et coll., 2015; Kumar et coll., 2017; Møller et coll., 2018).
Fait intéressant, dans un article récent, Møller et al. des milliers d’ECCDNA ont été identifiés dans les leucocytes et les cellules musculaires des témoins sains. Dans l’idée d’étudier si les ADNC peuvent être transcrits, une bibliothèque d’ARNm a également été séquencée à partir de tissus musculaires et analysée pour détecter les événements de transcription à travers la jonction du point d’arrêt de l’ADNc détecté, trouvant plusieurs correspondances (Møller et al., 2018). Cette découverte suggère que l’ADN circulaire dans les tissus sains est transcrit, donnant lieu à des transcrits linéaires et polyadénylés qui porteront une séquence équivalente à la séquence de rétrodiffusion des CIRCRNA (Møller et al., 2018) (Figure 2).
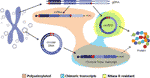
Figure 2 Représentation graphique de différents transcrits issus soit de l’ADN génomique, soit de l’ADN circulaire. Les exons sont colorés en violet et les jonctions dorsales ou les jonctions chimériques sont représentées en rouge. Les transcrits polyadénylés, contenant des jonctions chimériques et résistants à la RNase R sont surlignés en orange, bleu et jaune respectivement.
Les preuves transcriptionnelles des ADN circulaires, ainsi que leur abondance, nous amènent à suggérer que les ADN circulaires pourraient être une source naturelle d’une quantité importante d’ARN linéaires porteurs de jonctions chimériques. Dans de nombreux cas, ces jonctions chimériques peuvent être indiscernables des jonctions à rétrodiffusion des CIRCNAS, et par conséquent, elles peuvent être des facteurs de confusion dans les études sur les circNAS. Dans les paragraphes suivants, nous expliquerons les données à l’appui de cette proposition.
Détection de circRNA: Tout Ce Qui Brille N’Est Pas de l’Or
Comme précédemment introduit, les circRNAs sont formés par un événement d’épissage non canonique appelé backsplicing. Les transcrits résultant de cet événement de rétrodiffusion ont une structure en boucle fermée de manière covalente sans polarité 5′-3′, ni queue polyadénylée et, plus important encore, ils sont caractérisés par la présence d’un ordre d’exon brouillé par rapport au transcrit linéaire (Zhang et al., 2016; Wilusz, 2018). Cet ordre d’exon brouillé devient évident dans la jonction en revers qui relie une séquence aval 5′ à une séquence amont 3′. Ainsi, tous les algorithmes de détection de circRNA exploitent la présence des jonctions épissées en arrière comme caractéristique diagnostique pour l’identification de circRNA (Figure 1A).
Différentes méthodes ont été adaptées pour la détection de ces jonctions rétro-épissées. Des réseaux commerciaux contenant des sondes ciblant ces régions rétrodiffusées ont été largement utilisés dans les études de criblage de biomarqueurs (Iparraguirre et al., 2017; Liu et coll., 2017; Sui et coll., 2017; Li et coll., 2018). La validation ultérieure est souvent également basée sur l’amplification des jonctions de rétrodiffusion à l’aide d’amorces divergentes (Panda et Gorospe, 2018). De nombreux autres articles ont mené une analyse de séquençage à haut débit qui surmonte l’une des principales limitations des réseaux permettant de détecter non seulement les CIRCARNAS annotés, mais également les événements de circularisation de l’ARN de novo à partir de régions génomiques où aucun CIRCARNA n’a été annoté par des études précédentes. Plusieurs pipelines bioinformatiques ont été développés pour la détection des CIRCNAS dans les ensembles de données ARN-Seq, mais tous sont basés sur la présence de lectures croisant les jonctions d’épissage arrière et trouver la plus fiable reste un défi pour les bioinformaticiens (Hansen et al., 2016; Hansen, 2018; Prada-Luengo et al., 2019).
Deux approches principales peuvent être suivies pour la détection des CIRCRNA dans les données ARN-Seq. Tout d’abord, de nombreuses études d’ARN circRNA-seq sont basées sur des échantillons traités à la RNase R afin d’épuiser tous les ARN linéaires avant le séquençage. Bien que cette approche soit spécialement conçue pour la détection des circRNA, il convient de noter que la dégradation de la RNase R est variable, qu’il existe de rares cas d’ARN linéaires résistants à la RNase R et de CIRCRNA sensibles à la RNase R (Szabo et Salzman, 2016) et que ce traitement nécessite un apport d’ARN élevé qui pourrait être limitatif pour certains tissus. D’autres études sur les circNNA choisissent de séquencer des ARN totaux, appauvris en ribosomes (ribo-) ou non polyadénylés (polyA-), où des ARN linéaires et circulaires peuvent être trouvés (Salzman et al., 2012; Memczak et coll., 2013; Broadbent et coll., 2015; Lu et coll., 2015; Memczak et coll., 2015). Cette approche évite l’utilisation de la RNase R, qui réduit la quantité d’ARN nécessaire au séquençage et permet d’étudier l’expression d’autres types d’ARN à partir du même ensemble de données. Il a été démontré qu’avec une bonne profondeur et une bonne qualité de séquençage et une analyse minutieuse des données, de vrais CIRCARNAS peuvent être détectés à partir du séquençage total de l’ARN (Wang et al., 2017), cependant, dans cette deuxième approche, une confirmation de circularité ultérieure pour les CIRCRNA est nécessaire.
Avec la découverte d’ARN chimériques linéaires transcrits à partir d’ADN circulaires, les CIRCRNA ne sont plus les seuls transcrits avec des jonctions chimériques. Par conséquent, il est de la plus haute importance de noter que si la première approche enrichira significativement l’échantillon d’ARN dans les CIRCRNA de sorte que la plupart des jonctions chimériques détectées correspondent à des CIRCRNA vrais, la seconde pourrait surestimer le nombre de transcrits de circRNA en attribuant aux CIRCRNA le signal provenant à la fois des CIRCRNA et des transcrits chimériques linéaires transcrits à partir d’ADN circulaires. Par conséquent, compte tenu de la coexistence des circRNAs et des transcriptions chimériques linéaires, le besoin de tests de circularité et de tests de fonctionnalité gagne en importance et un soin particulier doit être pris en ce qui concerne non seulement les méthodes expérimentales mais aussi informatiques pour éviter de confondre les transcriptions chimériques des ADN circulaires avec des circRNAs formés par rétrodiffusion.
Discussion
Le champ des circRNA est encore à un stade précoce, cependant, les CIRCRNA se sont déjà révélés être des molécules étonnantes, impliquées dans de nombreux processus, avec un grand potentiel de biomarqueurs et qui peuvent également changer la façon dont nous comprenons les processus de transcription et de traduction. Pour ces raisons, ils attirent l’attention et le champ circRNA est actuellement l’un des champs de recherche sur l’ARN les plus actifs. Cependant, de nombreux conflits, controverses et questions ouvertes (Li, 2019) doivent encore être discutés.
Dans ce rapport, et à la lumière des progrès récents dans le domaine de l’ADN circulaire, nous voulons souligner la transcription à partir d’ADN circulaire extrachromosomique comme l’une des principales sources naturelles de transcriptions linéaires avec des signaux rétroprojecteurs qui pourraient interférer avec les données sur l’ARNC (Møller et al., 2018). Désormais, outre les artefacts techniques, les duplications et les événements de transplantation qui pourraient conduire à des faux positifs dans la détection des circNNA, nous devons également prendre en compte l’existence de ce nouveau type de transcriptions chimériques. Par conséquent, les tests de circularisation et les tests fonctionnels sont plus importants que jamais.
Dans tous les cas, ces transcriptions linéaires chimériques ne doivent pas seulement être considérées comme un simple facteur de confusion pour les études de circRNA. Malgré les implications techniques pour la caractérisation des ARNC, l’existence de ces molécules d’ARN chimériques linéaires semblables à des ARNC provenant d’ECCDNA ajoute un nouveau type de molécule à la liste toujours croissante des ARN et élargit notre vision de la complexité du transcriptome et de sa régulation. De plus, ces molécules d’ARN linéaires issues de l’eccDNA pourraient également présenter des fonctions similaires au circRNA, notamment les fonctions régulatrices ou le potentiel à traduire. Les produits géniques des transcrits de l’adnCEC pourraient potentiellement contribuer au phénotype des cellules et des tissus somatiques, tel que rapporté chez la levure (Gresham et al., 2010; Demeke et coll., 2015). Cependant, dans ce domaine naissant, davantage de données et de recherches sont nécessaires pour commencer à gratter la surface de l’iceberg.
Contributions des auteurs
LI, IP-L, BR et DO ont écrit le document.
Financement
Cette étude a été financée par l’Instituto de Salud Carlos III à travers le projet « PI17/00189 » (cofinancé par le Fonds Européen de Développement Régional / Fonds Social Européen) « Investir dans votre avenir »). IP-L et BR ont été soutenus par le Conseil Danois pour la Recherche indépendante, 6108-00171B et LI a été soutenu par le Ministère de l’Éducation du gouvernement basque.
Conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de relations commerciales ou financières pouvant être interprétées comme un conflit d’intérêts potentiel.