Holger J Schünemann, Julian PT Higgins, Gunn E Vist, Paul Glasziou, Elie A Akl, Nicole Skoetz, Gordon H Guyatt; au nom du Groupe des Méthodes de graduation Cochrane (anciennement Groupe des Méthodes d’Applicabilité et de Recommandations) et du Groupe des Méthodes Statistiques Cochrane
- Clé Points:
- 14.1 Tableaux » Résumé des résultats »
- 14.1.1 Introduction aux tableaux » Résumé des résultats »
- 14.1.2 Sélection des résultats pour les tableaux « Résumé des résultats »
- 14.1.3 Modèle général pour les tableaux de » Résumé des résultats »
- 14.1.4 Production de tableaux de » Synthèse des résultats »
- 14.1.5 Considérations statistiques dans les tableaux » Résumé des résultats »
- 14.1.5.1 Résultats dichotomiques
- 14.1.5.2 Résultats du délai jusqu’à l’événement
- 14.1.6 Contenu détaillé d’un tableau de « Synthèse des résultats »
- 14.1.6.1 Titre et en-tête du tableau
- 14.1.6.2 Résultats
- 14.1.6.3 Meilleure estimation du risque avec l’intervention du comparateur
- 14.1.6.4 Risque avec intervention
- 14.1.6.5 Différence de risque
- 14.1.6.6 Effet relatif (IC à 95 %)
- 14.1.6.7 Nombre de participants (études)
- 14.1.6.8 Certitude de la preuve (NOTE)
- 14.1.6.9 Commentaires
- 14.1.6.10 Explications
- 14.2 Évaluation de la certitude ou de la qualité d’un ensemble de preuves
- 14.2.1 L’approche par NOTES
Clé Points:
- Un tableau « Résumé des résultats » pour une comparaison donnée des interventions fournit des informations clés concernant l’ampleur des effets relatifs et absolus des interventions examinées, la quantité de preuves disponibles et la certitude (ou la qualité) des preuves disponibles.
- Les tableaux » Résumé des résultats » comprennent une ligne pour chaque résultat important (jusqu’à un maximum de sept). Les formats acceptés de tableaux » Résumé des résultats » et de tableaux interactifs » Résumé des résultats » peuvent être produits à l’aide du logiciel GRADEpro GDT de GRADE.
- Cochrane a adopté l’approche par GRADES (Évaluation, Élaboration et évaluation des recommandations) pour évaluer la certitude (ou la qualité) d’un ensemble de preuves.
- L’approche par NOTES spécifie quatre niveaux de certitude pour un ensemble de preuves pour un résultat donné : élevé, modéré, faible et très faible.
- Les évaluations de degré de certitude sont déterminées en tenant compte de cinq domaines : risque de biais, incohérence, caractère indirect, imprécision et biais de publication. Pour les preuves provenant d’études non randomisées et d’études rarement randomisées, les évaluations peuvent ensuite être améliorées en tenant compte de trois autres domaines.
Citez ce chapitre comme suit: Schünemann HJ, Higgins JPT, Vist GE, Glasziou P, Akl EA, Skoetz N, Guyatt GH. Chapitre 14 : Compléter les tableaux du « Résumé des constatations » et évaluer la certitude des preuves. Dans: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (éditeurs). Manuel Cochrane pour les Revues systématiques des Interventions version 6.2 (mise à jour en février 2021). Cochrane, 2021. Disponible sur www.formation.cochrane.org/handbook .
14.1 Tableaux » Résumé des résultats »
14.1.1 Introduction aux tableaux » Résumé des résultats »
Les tableaux » Résumé des résultats » présentent les principales conclusions d’un examen sous forme de tableaux transparents, structurés et simples. En particulier, ils fournissent des informations clés concernant la certitude ou la qualité des preuves (c.-à-d. la confiance ou la certitude dans la plage d’une estimation d’effet ou d’une association), l’ampleur de l’effet des interventions examinées et la somme des données disponibles sur les principaux résultats. Les examens Cochrane devraient inclure des tableaux de » Résumé des résultats » lors de la planification et de la publication, et devraient comporter au moins un tableau clé de » résumé des résultats » représentant les comparaisons les plus importantes. Certains examens peuvent inclure plus d’un tableau de « résumé des résultats « , par exemple si l’examen porte sur plus d’une comparaison majeure, ou inclut des populations sensiblement différentes qui nécessitent des tableaux distincts (par exemple parce que les effets diffèrent ou qu’il est important d’afficher les résultats séparément). Dans la Base de données Cochrane des revues systématiques (CDSR), le tableau principal du » Résumé des résultats » d’une revue apparaît au début, avant la section Contexte. D’autres tableaux de » Résumé des résultats » apparaissent entre les sections Résultats et Discussion.
14.1.2 Sélection des résultats pour les tableaux « Résumé des résultats »
La planification du tableau « Résumé des résultats » commence au début de l’examen systématique, avec la sélection des résultats à inclure dans: (i) l’examen; et (ii) le tableau » Résumé des résultats ». Il s’agit d’une étape cruciale que les auteurs de la revue doivent aborder avec soin.
Pour assurer la production d’informations utiles de manière optimale, les revues Cochrane commencent par élaborer une question d’examen et par énumérer tous les principaux résultats importants pour les patients et les autres décideurs (voir les chapitres 2 et 3). L’approche de NOTATION pour évaluer la certitude de la preuve (voir la section 14.2) définit et met en œuvre un processus de notation qui aide à séparer les résultats en ceux qui sont critiques, importants ou non importants pour la prise de décision. La consultation et la rétroaction sur le protocole d’examen, y compris de la part des consommateurs et d’autres décideurs, peuvent améliorer ce processus.
Les résultats critiques sont susceptibles d’inclure des paramètres clairement importants; des exemples typiques incluent la mortalité et la morbidité majeure (comme les accidents vasculaires cérébraux et l’infarctus du myocarde). Cependant, ils peuvent également représenter des effets secondaires mineurs et rares, des symptômes, une qualité de vie, des fardeaux associés au traitement et des problèmes de ressources (coûts). Les charges représentent l’impact de la charge de travail des soins de santé sur la fonction et le bien-être des patients, et comprennent les exigences d’adhérer à une intervention que les patients ou les soignants (par exemple, la famille) peuvent ne pas aimer, comme devoir subir des tests plus fréquents, ou les restrictions sur le mode de vie que certaines interventions nécessitent (Spencer-Bonilla et al 2017).
Fréquemment, lors de la formulation de questions qui incluent tous les résultats importants pour le patient pour la prise de décision, les auteurs de la revue confronteront les rapports d’études qui n’ont pas inclus tous ces résultats. Cela est particulièrement vrai pour les résultats défavorables. Par exemple, les essais randomisés pourraient apporter des preuves sur les effets escomptés et sur les effets secondaires fréquents et relativement mineurs, mais ne pas rapporter de résultats indésirables rares tels que les tentatives de suicide. Le chapitre 19 traite des stratégies de lutte contre les effets néfastes. Pour obtenir des données sur tous les résultats importants, il peut être nécessaire d’examiner les résultats d’études non randomisées (voir chapitre 24). Cochrane, en collaboration avec d’autres, a élaboré des lignes directrices à l’intention des auteurs de revues pour appuyer leur décision quant au moment de rechercher et d’inclure des études non randomisées (Schünemann et al 2013).
Si un examen ne comprend que des essais randomisés, ces essais peuvent ne pas aborder tous les résultats importants et il peut donc ne pas être possible de traiter ces résultats dans les limites de l’examen. Les auteurs de la revue doivent reconnaître ces limites et les rendre transparentes pour les lecteurs. Les auteurs de la revue sont encouragés à inclure des études non randomisées pour examiner les effets indésirables rares ou à long terme qui peuvent ne pas être étudiés de manière adéquate dans les essais randomisés. Cela soulève la possibilité que les résultats néfastes proviennent d’études dans lesquelles les participants diffèrent de ceux des études utilisées dans l’analyse des avantages. Les auteurs de la revue devront ensuite examiner dans quelle mesure ces différences sont susceptibles d’avoir une incidence sur les résultats, ce qui aura une incidence sur la certitude des preuves en raison des préoccupations concernant l’indirection liée à la population (voir la section 14.2.2).
Les études non randomisées peuvent fournir des informations importantes non seulement lorsque les essais randomisés ne rendent pas compte d’un résultat ou que les essais randomisés souffrent d’un caractère indirect, mais aussi lorsque les preuves issues des essais randomisés sont jugées très faibles et que les études non randomisées fournissent des preuves d’une certitude plus élevée. Un examen plus approfondi de ces questions figure également au chapitre 24.
14.1.3 Modèle général pour les tableaux de » Résumé des résultats »
Plusieurs versions standard alternatives des tableaux de » Résumé des résultats » ont été élaborées pour assurer la cohérence et la facilité d’utilisation entre les examens, l’inclusion des informations les plus importantes dont les décideurs ont besoin et une présentation optimale (voir les exemples à la figure 14.1.a et 14.1.b). Ces formats sont soutenus par des recherches axées sur une meilleure compréhension des informations qu’ils ont l’intention de transmettre (Carrasco-Labra et al 2016, Langendam et al 2016, Santesso et al 2016). Ils sont disponibles via le progiciel officiel de GRADE développé pour soutenir l’approche de GRADE: GRADEpro GDT (www.gradepro.org ).
Les tableaux standard du » Résumé des résultats » de Cochrane comprennent les éléments suivants en utilisant l’un des formats acceptés. La section 14.1.6 donne des indications supplémentaires sur chacun de ces aspects.
- Une brève description de la population et du milieu abordés par les données probantes disponibles (qui peuvent être légèrement différentes ou plus étroites que celles définies par la question d’examen).
- Une brève description de la comparaison abordée dans le tableau » Résumé des résultats « , y compris les interventions expérimentales et les interventions de comparaison.
- Une liste des résultats les plus critiques et/ou importants pour la santé, souhaitables et indésirables, limitée à sept résultats ou moins.
- Mesure du fardeau typique de chaque résultat (par exemple, risque illustratif, ou moyenne illustrative, sur une intervention de comparaison).
- L’amplitude absolue et relative de l’effet mesurée pour chacun (si les deux sont appropriés).
- Le nombre de participants et d’études contribuant à l’analyse de chaque résultat.
- Une évaluation GRADE de la certitude globale de l’ensemble des preuves pour chaque résultat (qui peut varier selon le résultat).
- Espace pour les commentaires.
- Explications (anciennement appelées notes de bas de page).
Idéalement, les tableaux du » résumé des constatations » sont étayés par des tableaux plus détaillés (appelés » profils de preuves « ) auxquels l’examen peut être lié, qui fournissent des explications plus détaillées. Les profils de données probantes comprennent les mêmes résultats importants pour la santé et fournissent plus de détails que les tableaux de « Résumé des résultats » des deux considérations individuelles qui alimentent le classement de la certitude et des résultats des études (Guyatt et al 2011a). Ils veillent à ce qu’une approche structurée soit utilisée pour évaluer la certitude de la preuve. Bien qu’ils soient rarement publiés dans les revues Cochrane, les profils de données probantes sont souvent utilisés, par exemple, par les concepteurs de lignes directrices pour examiner la certitude des données probantes à l’appui des recommandations des lignes directrices. Les auteurs de la revue trouveront plus facile d’élaborer le tableau « Résumé des constatations » en complétant d’abord l’évaluation de la certitude de la preuve dans le profil de la preuve dans GRADEpro GDT. Ils peuvent ensuite le convertir automatiquement dans l’un des formats de « Résumé des résultats » de GRADEpro GDT, y compris un « Résumé des résultats » interactif pour publication.
En tant que mesure de l’ampleur de l’effet pour les résultats dichotomiques, le tableau « Résumé des résultats » devrait fournir une mesure relative de l’effet (par exemple, rapport de risque, rapport de cotes, danger) et des mesures du risque absolu. Pour d’autres types de données, une seule mesure absolue (telle qu’une différence de moyenne pour les données continues) peut suffire. Il est important que l’ampleur de l’effet soit présentée de manière significative, ce qui peut nécessiter une certaine transformation du résultat d’une méta-analyse (voir également Chapitre 15, Section 15.4 et Section 15.5). Les examens comportant plus d’une comparaison principale devraient inclure un tableau » Résumé des résultats » distinct pour chaque comparaison.
Figure 14.1.a fournit un exemple de tableau » Résumé des résultats « . Figure 15.1.b fournit un format de rechange qui peut faciliter davantage la compréhension et l’interprétation des conclusions de l’examen par les utilisateurs. Les preuves évaluant différents formats suggèrent que le tableau « Résumé des résultats » devrait inclure une différence de risque comme mesure de l’effet absolu et que les auteurs devraient de préférence utiliser un format qui inclut une différence de risque.
Une description détaillée du contenu d’un tableau » Résumé des constatations » figure à la section 14.1.6.
Figure 14.1.un exemple de tableau » Résumé des résultats »
Résumé des résultats (pour la version interactive, cliquez ici)
|
Bas de compression par rapport à aucun bas de compression pour les personnes prenant de longs vols |
||||||
|
Patients ou population: toute personne effectuant un long vol (d’une durée supérieure à 6 heures) Paramètres: transport aérien international Intervention: stockages de compressionsa Comparaison: sans bas |
||||||
|
Résultats |
Risques comparatifs illustratifs* (IC à 95 %) |
Effet relatif (IC à 95%) |
Nombre de participants (études) |
Certitude de la preuve (GRADE) |
||
|
Risque assumé |
Risque correspondant |
|||||
|
Sans bas |
Avec des bas |
|||||
|
Thrombose veineuse profonde symptomatique (TVP) |
Voir commentaire |
Voir le commentaire |
Non estimable |
(9 études) |
Voir le commentaire |
0 les participants ont développé une TVP symptomatique dans ces études |
|
TVP sans symptômes |
Population à faible risqueb |
RR 0.10 (0.04 à 0.26) |
(9 études) |
⊕⊕⊕⊕ Haut |
||
|
10 par 1000 |
1 pour 1000 (0 à 3) |
|||||
|
Population à haut risqueb |
||||||
|
20 par 1000 |
2 pour 1000 (1 à 8) |
|||||
|
Thrombose veineuse superficielle |
13 par 1000 |
6 pour 1000 (2 à 15) |
RR 0.45 (0.18 à 1.13) |
(8 études) |
⊕⊕⊕◯ Moderatec |
|
|
Oedème Valeurs après le vol mesurées sur une échelle allant de 0, pas d’œdème, à 10, œdème maximal |
Le score moyen d’œdème variait entre les groupes témoins de 6 à 9 |
Le score moyen d’œdème dans les groupes d’intervention était en moyenne 4,7 inférieur (IC à 95% -4,9 à -4.5) |
(6 études) |
⊕⊕◯◯ Bas |
||
|
Embolie pulmonaire |
Voir le commentaire |
Voir le commentaire |
Non estimable |
(9 études) |
Voir le commentaire |
0 les participants ont développé une embolie pulmonaire dans ces études |
|
Mort |
Voir le commentaire |
Voir le commentaire |
Pas estimable |
(9 études) |
Voir le commentaire |
0 les participants sont morts dans ces études |
|
Effets indésirables |
Voir le commentaire |
Voir le commentaire |
Non estimable |
(4 études) |
Voir le commentaire |
La tolérabilité des bas a été décrite comme très bonne sans se plaindre d’effets secondaires dans 4 études. |
|
* La base du risque supposé est fournie dans notes de bas de page. Le risque correspondant (et son intervalle de confiance à 95%) est basé sur le risque supposé dans le groupe d’intervention et l’effet relatif de l’intervention (et son IC à 95%). IC : intervalle de confiance; RR: rapport de risque; GRADE : GRADES de preuves du Groupe de travail (voir explications). |
||||||
a Tous les bas des neuf études incluses dans cette revue étaient des bas de compression sous le genou. Dans quatre études, la force de compression était de 20 mmHg à 30 mmHg à la cheville. Il était de 10 mmHg à 20 mmHg dans les quatre autres études. Les bas sont de différentes tailles. Si un bas est trop serré autour du genou, il peut empêcher un retour veineux essentiel provoquant une accumulation de sang autour du genou. Les bas de compression doivent être correctement ajustés. Un bas trop serré pourrait couper la peau lors d’un long vol et potentiellement provoquer une ulcération et un risque accru de TVP. Certains bas peuvent être légèrement plus épais que le couvre-jambe normal et peuvent être potentiellement restrictifs avec une usure serrée des pieds. C’est une bonne idée de porter des bas autour de la maison avant de voyager pour assurer un bon ajustement confortable. Les participants mettent leurs bas deux à trois heures avant le vol dans la plupart des études. La disponibilité et le coût des bas peuvent varier.
b Deux études ont recruté des participants à haut risque définis comme ceux ayant des épisodes antérieurs de TVP, des troubles de la coagulation, une obésité sévère, une mobilité réduite due à des problèmes osseux ou articulaires, une maladie néoplasique au cours des deux années précédentes, de grosses varices ou, dans l’une des études, des participants de plus de 190 cm et de plus de 90 kg. L’incidence des sept études qui excluaient les participants à risque élevé était de 1.45% et l’incidence pour les deux études qui ont recruté des participants à haut risque (avec au moins un facteur de risque) était de 2,43%. Nous avons utilisé 10 et 30 pour 1000 pour exprimer différentes strates de risque, respectivement.
c L’intervalle de confiance ne croise aucune différence et n’exclut pas une légère augmentation.
d La mesure de l’œdème n’a pas été validée (caractère indirect du résultat) ou aveuglée par l’intervention (risque de biais).
e S’il y a très peu ou pas d’événements et que le nombre de participants est important, le jugement sur la certitude des preuves (en particulier les jugements sur l’imprécision) peut être basé sur l’effet absolu. Ici, la cote de certitude peut être considérée comme « élevée » si le résultat a été évalué de manière appropriée et que l’événement ne s’est pas produit chez 2821 participants étudiés.
f Aucune des autres études n’a rapporté d’effets indésirables, à l’exception de quatre cas de thrombose veineuse superficielle dans les varices de la région du genou qui ont été comprimées par le bord supérieur du bas dans une étude.
Figure 14.1.b Exemple de tableau alternatif « Résumé des résultats «
|
Résumé des résultats (pour une version interactive, cliquez ici): |
||||||
|
Probiotiques comparés à aucun probiotique en complément aux antibiotiques chez les enfants |
||||||
|
Patient ou population: enfants recevant des antibiotiques Paramètres: patients hospitalisés et ambulatoires Intervention: probiotiques Comparaison: pas de probiotiques |
||||||
|
Résultats Nombre de participants (études) |
Effets relatifs |
Effets absolus prévus* (IC à 95 %) |
Certitude de la preuve |
|||
|
Sans probiotiques |
Avec des probiotiques |
Différence |
||||
|
Incidence de la diarrhée : Dose probiotique 5 milliards d’UFC/jour Suivi: 10 jours à 3 mois Enfants < 5 ans |
Enfants < 5 ans |
⊕⊕⊕⊝ modéréb En raison du risque de biais |
Diminue probablement l’incidence de la diarrhée. |
|||
|
1474 (7 études) |
RR 0,41 (0,29 à 0.55) |
22.3% a |
8.9% (6.5 à 12.2) |
13.4% moins d’enfantsen (10,1 à 15 ans.8 moins) |
||
|
Enfants > 5 ans |
Enfants > 5 ans |
⊕⊕⊝⊝ lowb, c En raison du risque de biais et d’imprécision |
Peut diminuer l’incidence de la diarrhée. |
|||
|
624 (4 études) |
RR 0,81 (0,53 à 1.21) |
11.2% a |
9% (5.9 à 13.6) |
2.2% moins d’enfantsun (5,3 de moins à 2.4 plus) |
||
|
Effets indésirables Suivi : 10 à 44 jours 1575 (11 études) |
1.8% a |
2.3% (0.8 à 3.8) |
0.5% plus d’événements indésirables (1 de moins à 2 de plus) |
⊕⊕⊝⊝ lowf, g En raison du risque de biais et d’incohérence |
Il peut y avoir peu ou pas de différence dans les événements indésirables. |
|
|
Durée de la diarrhée Suivi: 10 jours à 3 mois 897 (5 études) |
La durée moyenne des diarrhées sans probiotiques était de 4 jours. |
0.6 moins de jours (1,18 à 0,02 moins de jours) |
⊕⊕⊝⊝ lowh, i En raison de l’imprécision et de l’incohérence |
Peut diminuer la durée de la diarrhée. |
||
|
Selles par jour Suivi: 10 jours à 3 mois 425 (4 études) |
La moyenne de selles par jour sans probiotiques était de 2,5 selles par jour. |
0.3 moins de selles par jour (0,6 à 0 de moins) |
⊕⊕⊝⊝ lowj, k En raison de l’imprécision et de l’incohérence |
Il peut y avoir peu ou pas de différence dans les selles par jour. |
||
|
* La base du risque dans le groupe témoin (par exemple, le risque médian du groupe témoin dans l’ensemble des études) est fournie dans les notes de bas de page. Le risque dans le groupe d’intervention (et son intervalle de confiance à 95%) est basé sur le risque supposé dans le groupe témoin et l’effet relatif de l’intervention (et son IC à 95 %). IC : intervalle de confiance; RR : rapport de risque. |
||||||
|
EXPLICATIONS a les estimations des risques du groupe témoin proviennent d’estimations regroupées des groupes témoins. Effet relatif basé sur l’analyse de cas disponible b Risque élevé de biais dû à une perte élevée au suivi. c L’imprécision due au peu d’événements et aux intervalles de confiance comprend des avantages ou des inconvénients appréciables. d Effets secondaires: éruption cutanée, nausées, flatulences, vomissements, augmentation des mucosités, douleurs thoraciques, constipation, troubles du goût et manque d’appétit. e Les risques ont été calculés à partir des différences de risque regroupées. f Risque élevé de biais. Seuls 11 des 16 essais ont fait état d’événements indésirables, suggérant un biais de déclaration sélective. g Incohérence grave. De nombreux agents probiotiques et doses ont été évalués parmi un nombre relativement restreint d’essais, ce qui limite notre capacité à tirer des conclusions sur l’innocuité des nombreux agents probiotiques et doses administrées. h Incohérence inexpliquée grave (grande hétérogénéité I2 = 79%, valeur P, estimations ponctuelles et intervalles de confiance varient considérablement). je grave imprécision. La limite supérieure de 0,02 jour de diarrhée en moins n’est pas considérée comme importante pour le patient. j Incohérence inexpliquée grave (grande hétérogénéité I2 = 78%, valeur P, estimations ponctuelles et intervalles de confiance varient considérablement). k Imprécision grave. L’intervalle de confiance à 95% ne comprend aucun effet et la limite inférieure de 0,60 selles par jour est d’une importance discutable pour le patient. |
||||||
14.1.4 Production de tableaux de » Synthèse des résultats »
Le logiciel du Groupe de travail sur les NOTES, GRADEpro GDT (www.gradepro.org ), y compris le manuel interactif de GRADE, est disponible pour aider les auteurs de revues à préparer des tableaux de » Résumé des résultats « . GRADEpro peut utiliser les données sur le risque du groupe de comparaison et l’estimation de l’effet (saisies par les auteurs de la revue ou importées à partir de fichiers générés dans RevMan) pour produire les effets relatifs et les risques absolus associés aux interventions expérimentales. En outre, il guide l’utilisateur à travers le processus d’évaluation des notes et produit un tableau qui peut être utilisé comme un tableau autonome dans une revue (y compris par importation directe dans des logiciels tels que RevMan ou intégration avec RevMan Web), ou un tableau interactif de « Résumé des résultats » (voir ressources d’aide dans GRADEpro).
14.1.5 Considérations statistiques dans les tableaux » Résumé des résultats »
14.1.5.1 Résultats dichotomiques
Les tableaux » Résumé des résultats » devraient inclure des mesures absolues et relatives de l’effet des résultats dichotomiques. Les ratios de risque, les rapports de cotes et les différences de risque sont des façons différentes de comparer deux groupes avec des données de résultats dichotomiques (voir Chapitre 6, section 6.4.1). En outre, il existe deux ratios de risque distincts, selon l’événement (par exemple, » oui » ou » non « ) qui fait l’objet de l’analyse (voir Chapitre 6, section 6.4.1.5). En présence d’un effet d’intervention non nul, toute variation d’une étude à l’autre dans le groupe de comparaison risque (p. ex. la variation du risque que l’événement se produise sans l’intervention d’intérêt, par exemple dans différentes populations) rend impossible que plus d’une de ces mesures soit vraiment la même dans chaque étude.
On a longtemps supposé en épidémiologie que les mesures relatives de l’effet sont plus cohérentes que les mesures absolues de l’effet d’un scénario à l’autre. Il existe des preuves empiriques à l’appui de cette hypothèse (Engels et al 2000, Deeks et Altman 2001, Furukawa et al 2002). Pour cette raison, les méta-analyses devraient généralement utiliser un rapport de risque ou un rapport de cotes comme mesure de l’effet (voir Chapitre 10, section 10.4.3). En conséquence, une seule estimation de l’effet relatif est probablement un résumé plus approprié qu’une seule estimation de l’effet absolu. Si un effet relatif est effectivement cohérent d’une étude à l’autre, les risques de groupes de comparaison différents auront des implications différentes pour le bénéfice absolu. Par exemple, si le ratio de risque est systématiquement égal à 0.75, l’intervention expérimentale réduirait alors un risque du groupe de comparaison de 80% à 60% dans le groupe d’intervention (une réduction du risque absolu de 20 points de pourcentage), mais réduirait également un risque du groupe de comparaison de 20% à 15% dans le groupe d’intervention (une réduction du risque absolu de 5 points de pourcentage).
Les tableaux » Résumé des résultats » sont construits autour de l’hypothèse d’un effet relatif cohérent. Il est donc important de considérer les implications de cet effet pour les différents risques du groupe de comparaison (ceux-ci peuvent être dérivés ou estimés à partir d’un certain nombre de sources, voir la section 14.1.6.3), ce qui peut nécessiter une évaluation de la certitude des preuves pour les preuves pronostiques (Spencer et al 2012, Iorio et al 2015). Pour tout risque de groupe de comparaison, il est possible d’estimer un risque de groupe d’intervention correspondant (c’est-à-dire le risque absolu avec l’intervention) à partir du rapport de risque méta-analytique ou du rapport de cotes. Notez que les chiffres fournis dans la colonne » Risque correspondant » sont spécifiques aux » risques » de la colonne adjacente.
Pour le rapport de risque méta-analytique (RR) et le risque de comparaison supposé (ACR), le risque d’intervention correspondant est obtenu comme suit ::
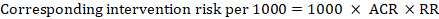 .
.
À titre d’exemple, dans la figure 14.1.a, le rapport de risque méta-analytique pour la thrombose veineuse profonde sans symptômes (TVP) est RR = 0,10 (IC à 95% de 0,04 à 0,26). En supposant un risque comparateur d’ACR = 10 pour 1000 = 0,01, on obtient:
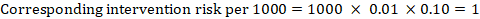 .
.
Pour le rapport de cotes méta-analytique (RO) et le risque de comparaison supposé, ACR, le risque d’intervention correspondant est obtenu comme suit ::
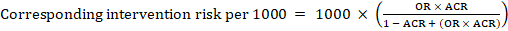 .
.
Les limites de confiance supérieure et inférieure pour le risque d’intervention correspondant sont obtenues en remplaçant RR ou OR par leurs limites de confiance supérieure et inférieure, respectivement (par exemple en remplaçant 0.10 avec 0,04, puis avec 0,26, dans l’exemple). Ces intervalles de confiance n’intègrent pas d’incertitude dans les risques de comparaison supposés.
Lorsqu’il s’agit de ratios de risque, il est essentiel d’utiliser la même définition d' » événement » que celle utilisée pour la méta-analyse. Par exemple, si la méta-analyse portait sur la » mort » (par opposition à la survie) comme événement, les risques correspondants dans le tableau « Résumé des résultats » doivent également faire référence à la » mort ».
Dans des circonstances (rares) où il est clairement justifié de supposer une différence de risque cohérente dans la méta-analyse, il est en principe possible de présenter cela pour les « risques assumés » pertinents et leurs risques correspondants, et de présenter les effets relatifs correspondants (différents) pour chaque risque assumé.
La différence de risque exprime la différence entre l’ACR et le risque d’intervention correspondant (ou la différence entre l’intervention expérimentale et l’intervention de comparaison).
Pour le rapport de risque méta-analytique (RR) et le risque de comparaison présumé (ACR), la différence de risque correspondante est obtenue comme suit (notez que les risques peuvent également être exprimés en pourcentage ou en points de pourcentage):
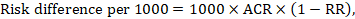
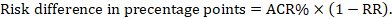
À titre d’exemple, à la figure 14.1.b le rapport de risque méta-analytique est de 0,41 (IC à 95% de 0,29 à 0,55) pour la diarrhée chez les enfants de moins de 5 ans. En supposant un risque de groupe de comparaison de 22,3%, nous obtenons:
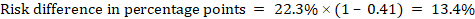 .
.
Pour le rapport de cotes méta-analytique (RO) et le risque de comparaison présumé (ACR), la différence de risque absolue est obtenue en (points de pourcentage):
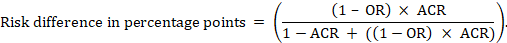
Les limites de confiance supérieure et inférieure pour la différence de risque absolue sont obtenues en réexécutant le calcul ci-dessus tout en remplaçant RR ou OR par leurs limites de confiance supérieure et inférieure, respectivement (par exemple en remplaçant 0,41 par 0,28, puis par 0,55, dans l’exemple). Ces intervalles de confiance n’intègrent pas d’incertitude dans les risques de comparaison supposés.
14.1.5.2 Résultats du délai jusqu’à l’événement
Les résultats du délai jusqu’à l’événement mesurent si et quand un événement particulier (par exemple, un décès) se produit (van Dalen et al., 2007). L’impact de l’intervention expérimentale par rapport au groupe témoin sur les résultats du temps à l’événement est généralement mesuré à l’aide d’un rapport de risque (HR) (voir Chapitre 6, section 6.8.1).
Un rapport de risque exprime une estimation de l’effet relatif. Il peut être utilisé de diverses manières pour obtenir des risques absolus et d’autres quantités interprétables pour une population spécifique. Nous décrivons ici comment exprimer à nouveau les rapports de risque en termes de: (i) risque absolu de survie sans événement dans une période donnée; (ii) risque absolu d’un événement dans une période donnée; et (iii) temps médian avant l’événement. Toutes les méthodes reposent sur l’hypothèse d’effets relatifs cohérents (c’est-à-dire que le rapport de risque ne varie pas dans le temps).
(i) Risque absolu de survie sans événement au cours d’une période donnée La survie sans événement (par exemple la survie globale) est généralement rapportée par des études individuelles. Pour obtenir des effets absolus pour les résultats du temps avant l’événement mesurés comme une survie sans événement, le résumé HR peut être utilisé en conjonction avec une proportion supposée de patients sans événement dans le groupe de comparaison (Tierney et al., 2007). Cette proportion de patients sera spécifique à une période de temps d’observation. Cependant, il n’est pas strictement nécessaire de spécifier cette période. Par exemple, une proportion de 50% des patients sans événement pourrait s’appliquer à des patients avec un taux d’événement élevé observé sur 1 an, ou à des patients avec un taux d’événement faible observé sur 2 ans.
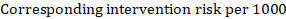
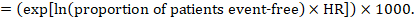
À titre d’exemple, supposons que le rapport de risque méta-analytique soit de 0,42 (IC à 95% de 0,25 à 0,72). En supposant un risque de survie sans événement du groupe de comparaison (par exemple pour les personnes vivant en survie globale) à 2 ans d’ACR = 900 pour 1000 = 0,9, nous obtenons:
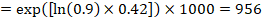
pour que 956 personnes pour 1000 soient vivantes avec l’intervention expérimentale à 2 ans. La dérivation du risque doit être expliquée dans un commentaire ou une note de bas de page.
(ii) Risque absolu d’un événement dans une période de temps donnée Pour obtenir cet effet absolu, le résumé HR peut à nouveau être utilisé (Tierney et al 2007):
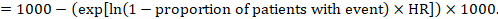
Dans l’exemple, supposons que nous supposons un risque d’événements du groupe de comparaison (par exemple pour la mortalité, les personnes décédées) à 2 ans d’ACR = 100 pour 1000 = 0,1. Nous obtenons:
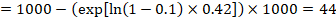
pour que 44 personnes sur 1000 soient mortes avec l’intervention expérimentale à 2 ans.
(iii) Temps médian avant l’événement Au lieu de chiffres absolus, le temps avant l’événement dans les groupes d’intervention et de comparaison peut être exprimé en temps de survie médian en mois ou en années. Pour obtenir le temps de survie médian, le HR mis en commun peut être appliqué à un temps de survie médian supposé dans le groupe de comparaison (Tierney et al. 2007):
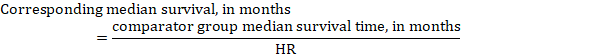
Dans l’exemple, en supposant un temps de survie médian du groupe de comparaison de 80 mois, on obtient:
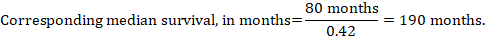
Pour ces trois options de ré-expression des résultats des analyses de temps à événement, les limites de confiance supérieure et inférieure pour le risque d’intervention correspondent sont obtenues en remplaçant HR par ses limites de confiance supérieure et inférieure, respectivement (par exemple en remplaçant 0,42 par 0,25, puis par 0,72, dans l’exemple). Encore une fois, comme pour les résultats dichotomiques, ces intervalles de confiance n’intègrent pas d’incertitude dans les risques supposés du groupe de comparaison. Ceci est particulièrement préoccupant pour la survie à long terme avec un taux de mortalité faible ou modéré et un nombre élevé de patients censurés correspondant (c’est-à-dire un faible nombre de patients à risque et un taux de censure élevé).
14.1.6 Contenu détaillé d’un tableau de « Synthèse des résultats »
14.1.6.1 Titre et en-tête du tableau
Le titre de chaque tableau de « Synthèse des résultats » doit préciser la question de santé, articulée en fonction de la population et préciser exactement la comparaison des interventions. Figure 14.1.r, la population est constituée de personnes prenant de longs vols en avion, l’intervention est des bas de compression et le contrôle n’est pas des bas de compression.
Les premières lignes de chaque tableau « Résumé des résultats » doivent fournir les informations d’en-tête suivantes:
Patients ou population Cela clarifie davantage la population (et éventuellement les sous-populations) d’intérêt et, idéalement, l’ampleur du risque de résultat indésirable le plus crucial auquel une intervention est dirigée. Par exemple, les personnes sur un vol long-courrier peuvent être exposées à différents risques de TVP; ceux qui utilisent des inhibiteurs sélectifs de la recapture de la sérotonine (ISRS) peuvent présenter un risque différent d’effets secondaires; tandis que ceux atteints de fibrillation auriculaire peuvent présenter un risque annuel d’AVC faible (< 1%), modéré (1% à 4%) ou élevé (> 4%).
Cette définition devrait indiquer toutes les caractéristiques spécifiques des paramètres de la question de santé qui pourraient limiter l’applicabilité du résumé des résultats à d’autres paramètres (par exemple, les soins primaires en Europe et en Amérique du Nord).
Intervention L’intervention expérimentale.
Comparaison L’intervention du comparateur (y compris aucune intervention spécifique).
14.1.6.2 Résultats
Les lignes d’un tableau » Résumé des résultats » devraient inclure tous les résultats souhaitables et indésirables pour la santé (classés par ordre d’importance) qui sont essentiels à la prise de décision, jusqu’à un maximum de sept résultats. S’il y a plus de résultats dans l’examen, les auteurs de l’examen devront omettre les résultats les moins importants du tableau, et la décision de choisir les résultats essentiels ou importants pour l’examen devrait être prise lors de l’élaboration du protocole (voir le chapitre 3). Les auteurs de la revue devraient fournir des délais pour la mesure des résultats (par exemple, 90 jours ou 12 mois) et le type de notes de l’instrument (par exemple, allant de 0 à 100).
Il est à noter que les auteurs de la revue devraient inclure les résultats critiques et importants préétablis dans le tableau, que les données soient disponibles ou non. Cependant, ils devraient être attentifs à la possibilité que l’importance d’un résultat (par exemple un effet négatif grave) ne soit connue qu’après la rédaction du protocole ou la réalisation de l’analyse, et devraient prendre les mesures appropriées pour les inclure dans le tableau « Résumé des résultats ».
Le tableau » Résumé des résultats » peut inclure les effets dans les sous-groupes de la population pour différents risques de comparaison et tailles d’effets séparément. Par exemple, à la figure 14.1.les effets b sont présentés séparément pour les enfants de moins de 5 ans et de plus de 5 ans. Les auteurs de la revue peuvent également choisir de produire des tableaux distincts de » Résumé des résultats » pour différentes populations.
Les auteurs de la revue devraient inclure les événements indésirables graves, mais il pourrait être possible de combiner les événements indésirables mineurs en un seul résultat, et de le décrire dans une note de bas de page explicative (notez qu’il n’est pas approprié d’additionner les événements à moins qu’ils ne soient indépendants, c’est-à-dire qu’un participant qui a subi un événement indésirable n’a aucune chance de subir l’autre événement indésirable).
Les résultats mesurés à plusieurs moments représentent un problème particulier. En général, pour simplifier le tableau, les auteurs de la revue ne devraient présenter plusieurs points temporels que pour les résultats essentiels à la prise de décision, où le résultat ou la décision prise sont susceptibles de varier au fil du temps. Le reste devrait être présenté à un moment commun si possible.
Les auteurs de la revue peuvent présenter des mesures continues des résultats dans le tableau » Résumé des résultats » et devraient s’efforcer de les rendre interprétables pour le public cible. Cela nécessite que les unités soient claires et facilement interprétables, par exemple les jours de douleur ou la fréquence des maux de tête, et que le nom et l’échelle des outils de mesure utilisés soient indiqués (par exemple une échelle analogique visuelle, allant de 0 à 100). Cependant, de nombreux instruments de mesure ne sont pas facilement interprétables par des cliniciens non spécialisés ou des patients, par exemple, des points sur un inventaire de dépression de Beck ou un score de qualité de vie. Pour ceux-ci, une présentation plus interprétable pourrait impliquer la conversion d’un résultat continu en un résultat dichotomique, tel qu’une amélioration > 50% (voir Chapitre 15, section 15.5).
14.1.6.3 Meilleure estimation du risque avec l’intervention du comparateur
Les auteurs de la revue devraient fournir jusqu’à trois risques typiques pour les participants recevant l’intervention du comparateur. Pour les résultats dichotomiques, nous recommandons que ceux-ci soient présentés sous la forme du nombre de personnes vivant l’événement pour 100 ou 1000 personnes (fréquence naturelle) en fonction de la fréquence du résultat. Pour les résultats continus, cela serait indiqué comme une valeur moyenne ou médiane du résultat mesuré.
Les risques estimés ou supposés d’intervention de comparaison pourraient être basés sur des évaluations des risques typiques dans différents groupes de patients dérivées de l’examen lui-même, d’études représentatives individuelles dans l’examen, ou de risques dérivés d’un examen systématique des études de pronostic ou d’autres sources de preuves qui peuvent à leur tour nécessiter une évaluation de la certitude des preuves pronostiques (Spencer et al 2012, Iorio et al 2015). Idéalement, les risques refléteraient des groupes que les cliniciens peuvent facilement identifier sur la base de leurs caractéristiques de présentation.
Une note explicative devrait préciser la source ou la justification de chaque risque de groupe de comparaison, y compris la période à laquelle il correspond, le cas échéant. Figure 14.1.r, les cliniciens peuvent facilement différencier les personnes présentant des facteurs de risque de thrombose veineuse profonde de celles qui n’en ont pas. S’il est connu qu’il y a peu de variation du risque de référence, les auteurs de l’examen peuvent utiliser le risque médian du groupe de comparaison entre les études. Si les risques typiques ne sont pas connus, une option consiste à choisir le risque parmi les études incluses, en fournissant le deuxième plus élevé pour une population à risque élevé et le deuxième plus faible pour une population à risque faible.
14.1.6.4 Risque avec intervention
Pour les résultats dichotomiques, les auteurs de la revue devraient fournir un risque absolu correspondant pour chaque risque de groupe de comparaison, ainsi qu’un intervalle de confiance. Ce risque absolu avec l’intervention (expérimentale) sera généralement dérivé du résultat de la méta-analyse présenté dans la colonne effet relatif (voir section 14.1.6.6). Les formules sont fournies à la section 14.1.5. Les auteurs de la revue doivent présenter l’effet absolu dans le même format que les risques liés à l’intervention du comparateur (voir section 14.1.6.3), par exemple en tant que nombre de personnes ayant vécu l’événement pour 1000 personnes.
Pour des résultats continus, une différence de moyenne ou une différence standardisée de moyenne doit être présentée avec son intervalle de confiance. Ceux-ci seront généralement obtenus directement à partir d’une méta-analyse. Un texte explicatif devrait être utilisé pour clarifier la signification, comme dans les figures 14.1.a et 14.1.b.
14.1.6.5 Différence de risque
Pour les résultats dichotomiques, la différence de risque peut être fournie en utilisant l’un des formats de tableau » Résumé des résultats » comme option supplémentaire (voir la figure 14.1.b). Cette différence de risque exprime la différence entre l’intervention expérimentale et l’intervention de comparaison et sera généralement dérivée du résultat de la méta-analyse présenté dans la colonne effet relatif (voir section 14.1.6.6). Les formules sont fournies à la section 14.1.5. Les auteurs de la revue doivent présenter la différence de risque dans le même format que les risques supposés et correspondants avec l’intervention du comparateur (voir la section 14.1.6.3); par exemple, en tant que nombre de personnes ayant vécu l’événement pour 1000 personnes ou en points de pourcentage si les risques supposés et correspondants sont exprimés en pourcentage.
Pour les résultats continus, si le tableau » Résumé des résultats » inclut cette option, la différence moyenne peut être présentée ici et la colonne » risque correspondant » laissée vide (voir Figure 14.1.b).
14.1.6.6 Effet relatif (IC à 95 %)
L’effet relatif sera généralement un rapport de risque ou un rapport de cotes (ou parfois un rapport de risque) avec l’intervalle de confiance de 95% qui l’accompagne, obtenu à partir d’une méta-analyse effectuée sur la base de la même mesure d’effet. Les ratios de risque et les rapports de cotes sont similaires lorsque les risques d’intervention des comparateurs sont faibles et que les effets sont faibles, mais peuvent différer considérablement lorsque les risques du groupe de comparaison augmentent. La méta-analyse peut impliquer une hypothèse d’effets fixes ou aléatoires, selon ce que les auteurs de la revue jugent approprié, et implique que l’effet relatif est soit une estimation de l’effet de l’intervention, soit une estimation de l’effet moyen de l’intervention dans l’ensemble des études, respectivement.
14.1.6.7 Nombre de participants (études)
Cette colonne devrait inclure le nombre de participants évalués dans les études incluses pour chaque résultat et le nombre correspondant d’études qui ont contribué à ces participants.
14.1.6.8 Certitude de la preuve (NOTE)
Les auteurs de la revue doivent commenter la certitude de la preuve (aussi appelée qualité de l’ensemble de preuves ou confiance dans les estimations des effets). Les auteurs de la revue doivent utiliser le système de notation spécifique des preuves développé par le Groupe de travail sur les NOTES (Atkins et al 2004, Guyatt et al 2008, Guyatt et al 2011a), qui est décrit en détail à la section 14.2. L’approche par NOTES classe la certitude dans un ensemble de preuves comme étant » élevée « , » modérée « , » faible » ou » très faible » par résultat. C’est le résultat d’un jugement, mais le processus de jugement fonctionne dans une structure transparente. À titre d’exemple, la certitude serait » élevée » si le résumé portait sur plusieurs essais randomisés présentant un faible risque de biais, mais la cote de certitude devient plus faible s’il y a des préoccupations au sujet du risque de biais, d’incohérence, de caractère indirect, d’imprécision ou de biais de publication. Les jugements autres que de « haute » certitude devraient être rendus transparents en utilisant des notes de bas de page explicatives ou la colonne « Commentaires » du tableau « Résumé des constatations » (voir la section 14.1.6.10).
14.1.6.9 Commentaires
L’objectif du champ « Commentaires » est d’aider à interpréter les informations ou les données identifiées dans la ligne. Par exemple, cela peut être sur la validité de la mesure des résultats ou la présence de variables associées à l’ampleur de l’effet. Les mises en garde importantes concernant les résultats doivent être signalées ici. Toutes les lignes n’auront pas besoin de commentaires, et il est préférable de laisser un blanc s’il n’y a rien qui justifie un commentaire.
14.1.6.10 Explications
Des explications détaillées doivent être incluses dans les notes de bas de page pour étayer les jugements du tableau » Résumé des résultats « , comme l’évaluation générale de la NOTE. Les explications doivent décrire la justification des aspects importants du contenu. Tableau 14.1.a énumère des conseils pour des explications utiles. Les explications doivent être concises, informatives, pertinentes, faciles à comprendre et précises. Si les explications ne peuvent pas être suffisamment décrites dans les notes de bas de page, les auteurs de la revue devraient fournir de plus amples détails sur les questions dans les sections Résultats et Discussion de la revue.
Tableau 14.1.un guide pour fournir des explications utiles dans les tableaux « Résumé des résultats » (SoF). Adapté de Santesso et al (2016)
Directives générales
- Entrez si possible les informations destinées aux lecteurs directement dans le tableau (par exemple, des informations sur la durée du suivi ou l’échelle utilisée).
- En règle générale, ne citez pas de références dans la section explications, sauf s’il existe des raisons spécifiques, par exemple, de fournir des informations sur les sources des risques de référence (voir point 3).
- Fournir la source d’information sur les risques de référence utilisés pour calculer les effets absolus.
- À la fin du tableau, examinez toutes les explications pour déterminer si certaines pourraient être référencées plusieurs fois si elles étaient reformulées ou combinées.
- Fournissez les raisons de la mise à niveau et de la rétrogradation des preuves (voir les directives spécifiques au domaine ci-dessous) et utilisez le logiciel GRADEpro GDT pour respecter les directives de NOTATION.
- L’ensemble des preuves d’un résultat particulier peut être déterminé comme présentant des problèmes graves ou très graves pour le domaine affecté (ou d’une gravité critique pour le risque de biais lorsque ROBINS-I est utilisé). Ainsi, il peut être utile d’indiquer le nombre de niveaux à déclasser (par exemple, déclassé d’un niveau pour le risque de biais), mais d’éviter la répétition de ce qui est dans le tableau (et l’impression de rapports formulés ou algorithmiques). Dans les profils de preuves, ces informations sont déjà dans les cellules du tableau.
- Bien que des explications sur la certitude de la preuve soient principalement nécessaires lorsqu’elles modifient la certitude, envisagez d’ajouter une explication lorsque la certitude de la preuve n’a pas été modifiée, mais lorsque cette décision peut être remise en question par d’autres. Cela aidera à comprendre les raisons du désaccord.
- Assurez-vous que le tableau ne sert pas à décrire les méthodes de l’examen (par exemple, ne décrivez pas les raisons de l’analyse statistique).
- Entrez les résultats pour les résultats qui n’ont pas pu être combinés statistiquement dans une méta-analyse (p. ex. résultats narratifs) directement dans le tableau SoF dans les colonnes des résultats. Une explication peut ne pas être nécessaire pour communiquer ces résultats. S’ils sont considérés comme bénéfiques pour le public visé, ajoutez des estimations complémentaires des effets de l’intervention (par exemple, le nombre nécessaire pour traiter les avantages et les inconvénients, la différence de risque exprimée en pourcentage, le résultat continu exprimé en unités de différence minimale importante) dans la colonne des commentaires.
- Utilisez les informations présentées dans les explications sur le processus de NOTATION pour éclairer d’autres parties clés de l’examen, y compris les versions sommaires et la discussion.
Guide spécifique au domaine pour la rédaction d’explications utiles
Risque de biais
- Décrivez le nombre d’études, ou la quantité d’informations qu’elles fournissent dans la méta-analyse, qui présentaient un risque élevé de biais et pour quel critère.
- Utilisez des termes tels que majorité, minorité, tout, une partie ou aucune; ou le nombre d’études comme études X/X.
- Pour les essais randomisés, mentionnez les critères spécifiques, y compris la dissimulation de la séquence d’allocation, la déclaration sélective des résultats, etc. Pour les études non randomisées, décrivez le critère dans l’outil utilisé (par exemple, en utilisant l’outil ROBINS-I).
- Indiquer si l’effet du risque de biais a été examiné dans une analyse de sensibilité. Le cas échéant, mentionnez la contribution des études présentant un risque élevé de biais aux estimations.
- Des informations sur la conception de l’étude peuvent être incluses dans les explications, en particulier dans les SoF lorsque différents plans d’étude sont inclus.
Incohérence
- Indiquez la mesure utilisée pour juger de l’incohérence, telle que le test ou la mesure statistique (I2, Chi2, Tau), ou le chevauchement des intervalles de confiance, ou la similitude des estimations ponctuelles.
- Si l’incohérence est basée sur I2, décrivez-la comme étant considérable, substantielle, modérée ou non importante.
- Le cas échéant, mentionnez si l’hétérogénéité a été explorée dans les analyses de sous-groupes par PICO (patients, intervention, comparaison, résultat) et s’il existe d’autres raisons potentielles à l’hétérogénéité.
- Dans le cas d’une seule étude pour un résultat, dites qu’il n’y a « aucun » plutôt que « sans objet ».
Imprécision
- Indiquer où la taille de l’échantillon ou le nombre d’événements ne correspond pas à la taille d’information optimale telle que calculée, ou aux « règles empiriques » (par exemple, 400 événements). Évitez de mentionner le nombre d’études comme motif d’imprécision.
- Indiquez si les intervalles de confiance incluent la possibilité d’un effet faible ou nul ET un avantage ou un préjudice important. Si elle est connue, indiquez la valeur numérique du seuil d’avantage important.
- Évitez de déclarer le résultat comme statistiquement ou non statistiquement significatif.
La mise à niveau
- Mentionne la raison de la mise à niveau : en raison d’un effet important, d’un gradient dose-réponse ou d’une confusion opposée résiduelle plausible augmente la certitude des preuves.
- Pour les effets importants, signalez si l’effet relatif est > 2 ou > 5. Pour les gradients dose-réponse, indiquez le niveau d’intervention et l’effet sur le résultat. Pour le domaine » confusion opposée résiduelle plausible « , décrivez l’effet du facteur de confusion sur l’estimation.
14.2 Évaluation de la certitude ou de la qualité d’un ensemble de preuves
14.2.1 L’approche par NOTES
Le Groupe de travail sur les notes de Recommandation, d’évaluation, de développement et d’évaluation (Groupe de travail sur les NOTES) a mis au point un système de notation de la certitude des preuves (Schünemann et al 2003, Atkins et al 2004, Schünemann et al 2006, Guyatt et al 2008, Guyatt et al 2011a). Plus de 100 organisations, dont l’Organisation mondiale de la Santé (OMS), l’American College of Physicians, l’American Society of Hematology (ASH), l’Agence canadienne des médicaments et de la Technologie de la Santé (ACMTS) et les National Institutes of Health and Clinical Excellence (NICE) au Royaume-Uni, ont adopté le système de notation (www.gradeworkinggroup.org ).
Cochrane a également officiellement adopté cette approche, et tous les examens Cochrane devraient utiliser le GRADE pour évaluer la certitude des preuves pour des résultats importants (voir Encadré MECIR 14.2.un).
Case MECIR 14.2.a Attentes pertinentes pour la conduite des examens d’intervention
|
C74 : Évaluation de la certitude de l’ensemble des preuves (obligatoire) |
|
|
Utilisez les cinq critères de NOTATION (risque de partialité, cohérence de l’effet, imprécision, caractère indirect et biais de publication) pour évaluer la certitude de l’ensemble des éléments de preuve pour chaque résultat et pour tirer des conclusions sur la certitude des éléments de preuve dans le texte de l’examen. |
La NOTE est l’approche la plus utilisée pour résumer la confiance dans les effets des interventions par résultat d’une étude à l’autre. Il est préférable d’utiliser l’outil GRADEpro en ligne, et de l’utiliser comme décrit dans le système d’aide du logiciel. Cela devrait aider à garantir que les équipes d’auteurs accèdent aux mêmes informations pour éclairer leurs jugements. Idéalement, deux personnes travaillant de manière indépendante devraient évaluer la certitude de l’ensemble des preuves et parvenir à un point de vue consensuel sur toute décision de déclassement. Les considérations relatives aux cinq GRADES doivent être prises en compte, que l’examen comporte ou non un tableau » Résumé des résultats « . Il est utile de tirer parti de ces informations dans la Discussion, dans les conclusions des auteurs et de transmettre la certitude des preuves dans le résumé abstrait et en langage clair. |
|
C75 : Justification des évaluations de la certitude de l’ensemble des preuves (obligatoire) |
|
|
Justifier et documenter toutes les évaluations de la certitude de l’ensemble des preuves (par exemple, déclassement ou mise à niveau en utilisant la NOTE). |
L’adoption d’une approche structurée garantit la transparence dans la formulation d’une interprétation des preuves, et le résultat est plus informatif pour l’utilisateur. |
Pour les examens systématiques, l’approche par GRADE définit la certitude d’un ensemble de preuves comme la mesure dans laquelle on peut être sûr qu’une estimation de l’effet ou de l’association est proche de la quantité d’intérêt spécifique. L’évaluation de la certitude d’un ensemble de preuves implique la prise en compte du risque de biais à l’intérieur et à l’intérieur de l’étude (limites de la conception et de l’exécution de l’étude ou qualité méthodologique), de l’incohérence (ou de l’hétérogénéité), du caractère indirect des preuves, de l’imprécision des estimations des effets et du risque de biais de publication (voir la section 14.2.2), ainsi que des domaines qui peuvent accroître notre confiance dans l’estimation des effets (comme décrit à la section 14.2.3). Le système de NOTATION implique une évaluation de la certitude d’un ensemble de preuves pour chaque résultat individuel. Les jugements sur les domaines qui déterminent la certitude de la preuve doivent être décrits dans la section résultats ou discussion et dans le tableau » Résumé des constatations « .
L’approche par GRADES spécifie quatre niveaux de certitude (figure 14.2.un). Pour les interventions, y compris les tests diagnostiques et autres qui sont évalués comme des interventions (Schünemann et al 2008b, Schünemann et al 2008a, Balshem et al 2011, Schünemann et al 2012), le point de départ pour évaluer la certitude des preuves est classé en deux types:
- essais randomisés; et
- études d’interventions non randomisées (IRSN), y compris les études d’observation (y compris, mais sans s’y limiter, les études de cohorte, les études cas-témoins, les études transversales, les séries de cas et les rapports de cas, bien que tous ces modèles ne soient généralement pas inclus dans les revues Cochrane).
Il existe de nombreux cas où les auteurs de la revue s’appuient sur les informations de l’IRSN, en particulier pour évaluer les dommages potentiels (voir le chapitre 24). En outre, les auteurs de la revue peuvent obtenir des données pertinentes à la fois des essais randomisés et du NRSI, chaque type de preuve complétant l’autre (Schünemann et al 2013).
En GRADE, un ensemble de preuves provenant d’essais randomisés commence par une cote de certitude élevée tandis qu’un ensemble de preuves provenant de l’IRSN commence par une cote de certitude faible. La cote inférieure avec le NRSI est le résultat du biais potentiel induit par l’absence de randomisation (c’est-à-dire le biais de confusion et de sélection).
Cependant, lors de l’utilisation du nouvel outil de Risque de biais Dans les études d’interventions Non randomisées (ROBINS-I) (Sterne et al 2016), un outil d’évaluation qui couvre le risque de biais dû à l’absence de randomisation, toutes les études peuvent commencer avec une grande certitude des preuves (Schünemann et al 2018). L’approche consistant à commencer tous les plans d’étude (y compris l’IRSN) en tant que certitude élevée n’entre pas en conflit avec l’approche initiale consistant à commencer l’évaluation de l’IRSN en tant que preuve de faible certitude. En effet, un ensemble de données probantes provenant de l’IRSN devrait généralement être déclassé de deux niveaux en raison du risque inhérent de biais associé à l’absence de randomisation, à savoir le biais de confusion et le biais de sélection. Le fait de ne pas rétrograder l’IRSN de certitude élevée à faible nécessite une justification transparente et détaillée de ce qui atténue les préoccupations concernant la confusion et le biais de sélection (Schünemann et al 2018). Il existe actuellement très peu d’exemples où il n’est pas approprié de réduire la note de deux niveaux.
La cote de certitude la plus élevée est un ensemble de preuves lorsqu’il n’y a aucune préoccupation concernant l’un des facteurs de NOTATION énumérés à la figure 14.2.a. Les auteurs de la revue rétrogradent souvent les preuves en preuves à certitude modérée, faible ou même très faible, en fonction de la présence des cinq facteurs de la figure 14.2.a. Habituellement, la cote de certitude diminue d’un niveau pour chaque facteur, jusqu’à un maximum de trois niveaux pour tous les facteurs. S’il existe des problèmes très graves pour n’importe quel domaine (p. ex. lors de l’évaluation du risque de biais, toutes les études n’ont pas été révélées, n’ont pas été aveuglées et ont perdu plus de 50% de leurs patients à suivre), les preuves peuvent chuter de deux niveaux en raison de ce seul facteur. Il n’est pas possible d’évaluer des preuves inférieures à une « très faible certitude ».
Les auteurs de la revue évalueront généralement les preuves provenant d’études non randomisées solides comme étant de faible certitude, même si ROBINS-I est utilisé. Si, toutefois, de telles études produisent des effets importants et qu’il n’y a pas de biais évident expliquant ces effets, les auteurs de la revue peuvent évaluer les preuves comme étant modérées ou – si l’effet est suffisamment important – même comme étant à haute certitude (figure 14.2.un). Le niveau de certitude très faible est approprié, sans s’y limiter, pour les études présentant des problèmes critiques et des observations cliniques non systématiques (par exemple, des séries de cas ou des rapports de cas).