introduktion
Limb-girdles Muskeldystrophies (LGMDs) inkluderer en heterogen gruppe af lidelser, der er kendetegnet ved den progressive spild og svaghed i de proksimale lemmer-girdles muskler (1, 2). Lgmd ‘ er viser Inter-og intrafamilial variabilitet, der spænder fra meget milde former til svær, tidlig begyndelse, hurtigt progressive fænotyper (2). Lgmd ‘ er kan klassificeres som autosomal dominant (LGMD1) eller recessiv (LGMD2). Den første gruppe har normalt en voksen alder og er ikke særlig almindelig (<10% af alle Lgmd ‘ er), mens sidstnævnte er hyppigere (1:15000) (1). Blandt recessive former er LGMD2A (eller calpainopati) den mest almindelige LGMD på verdensplan og påvirker cirka 30% af alle LGMDs-tilfælde (3). Kliniske signaturer af sygdom omfatter tiptoe gang, vraltende gangart, vanskeligheder med at løbe og klatre trapper, scapular vinger. Fælles kontrakturer, akillessenen forkortelse, skoliose observeres almindeligvis, mens ansigts-og nakke muskler ikke påvirkes (4). En asymptomatisk Hyperckæmi (5-80 gange CPK normale niveauer) hos unge patienter betragtes som et præklinisk sygdomsstadium og kan vedvare i flere år (1, 5). Alderen for muskelsvaghed forekommer normalt ved 15 år, selvom den kan opstå tidligere (<12 år) eller senere (>30 år) i alderen. Sygdomsprogressionen kan føre til tab af ambulation, respirationsinsufficiens og nedsat lunges vitale kapacitet i de avancerede stadier (6). Hjerteinddragelse (hjerterytmeforstyrrelser, hjerteledningsforstyrrelser, dysfunktion i venstre ventrikulær udstødning) rapporteres kun lejlighedsvis (6, 7).
diagnosen calpainopati bekræftes ved påvisning af patogene mutationer i CAPN3 (15k15.1) (4), der koder for forskellige alternativt splejsede transkripter. Imidlertid udtrykkes transkriptet i fuld længde primært i muskelvæv (8). Det kodede protein (CAPN3) er medlem af ikke-lysosomal Ca++-afhængig cysteinproteaser familie. I muskel deltager CAPN3 i” sarkomere remodeling”, som er afgørende for muskeltilpasning og vækst som reaktion på funktionelle og metaboliske krav. Til dato er mere end 490 patogene mutationer i hele CAPN3 blevet beskrevet, hvoraf de fleste er enkeltnukleotidændringer (4). Mutationer i CAPN3 har været forbundet med mitokondrie abnormiteter, vækstsvigt, øget oksidativ stress og sarkomer disorganisering, som helt bidrager til at gøre muskelen ude af stand til at holde belastninger, hvilket derved forårsager myofiber degeneration og muskelsvind (8). Diagnosen af calpainopati kan være udfordrende på grund af den genetiske heterogenitet og ikke-specificiteten af klinisk og instrumentelt mønster. Faktisk deles fordeling af muskelsvaghed/atrofi, hypertrofi/pseudo-hypertrofi og senekontrakturer meget ofte med andre Lgmd ‘ er eller neuromuskulære lidelser. Muskelbiopsi mønster i calpainopati er generelt ikke-specifikt også, lige fra milde muskulære abnormiteter til alvorlige dystrofiske ændringer. Desuden er immunhistokemiske / biokemiske markører normalt ikke pålidelige: calpain-signalet kan være normalt selv i nærvær af et ikke-funktionelt protein, og omvendt kan det reduceres selv i andre muskeldystrofier, der er forskellige fra calpainopati.
det anbefales derfor at udføre en differentieret diagnose for at give nøjagtige og pålidelige resultater. Til dette formål er molekylære genetiske testmetoder blevet udviklet til at bekræfte diagnosen calpainopati, herunder multigenpanel, omfattende genomisk (eksom/genomsekventering) og enkeltgenanalyse (direkte sekventering) (9). Implementeringen af næste generations sekventering (NGS) var nyttigt til at generere informative data, der skulle anvendes til diagnostiske, forudsigelige eller terapeutiske formål (10-12). NGS genpaneler er baseret på analysen af et sæt gener forbundet med en specifik sygdom eller en gruppe af relaterede lidelser, som er karakteriseret ved genetisk og fænotypisk heterogenitet. NGS-paneler kan også være nyttige til påvisning af blandede eller komplekse fænotyper, som skyldes arv af mere end en genetisk defekt fra forældre (13). Generelt er patienter, der præsenterer komplekse fænotyper, som er negative for de kendte mutationer i specialdesignede diagnostiske genpaneler og kræver en bred differentieret diagnose, berettiget til hel eksom-eller genomsekventering. Hele eksom/genomsekventering er dyrere tilgange med hensyn til datastyring, fortolkning og analytiske omkostninger, men øger sandsynligheden for at stille en molekylær diagnose til en mistænkt genetisk lidelse (13). Med hensyn til Lgmd ‘ er anbefales dedikerede NGS-paneler stærkt for at sikre høje diagnostiske hastigheder, optimal dækning, følsomhed og specificitet af klinisk test (9). NGS-paneler repræsenterer derfor et af de bedste systemer til at lette differentiel diagnose, identificere nye årsagsmutationer og afklare genotype-fænotype-korrelationer (14, 15).
i dette manuskript rapporteres sagen om en patient, der er påvirket af LGMD2A, for hvilken anvendelsen af NGS-panelet ikke kun tillod at bekræfte diagnosen calpainopati (mutationer i CAPN3), men også at identificere en yderligere, ny mutation i LMNA-gen forbundet med dilateret kardiomyopati. I betragtning af disse resultater blev analysen udvidet til familiemedlemmerne i Probanden for at give en mere omfattende fortolkning af de analytiske data i forhold til de patologiske fænotyper.
Case præsentation
klinisk karakterisering af Proband og slægtninge
sygdomsbegyndelsen i proband blev henvist til 10 år, da hun rapporterer tilstedeværelsen af kalvhypertrofi med tiptoe gang, vanskeligheder med at løbe, klatre trapper og stå op. Symptomerne viste en langsom, men progressiv forværring med efterfølgende involvering af proksimal øvre lem. I en alder af 13 blev høje niveauer af CPK (~8.000 UI/L) tilfældigt opdaget, aldrig forbundet med myoglobinuri. Successivt gennemgik patienten flere neuromuskulære undersøgelser på forskellige specialiserede centre. To muskelbiopsier blev udført, der viste klassisk dystrofisk billede (hypertrofiske/atrofiske fibre, indre kerner, nekrotiske fibre, stigning i bindevæv) med ikke-specifikke egenskaber. Som forventet var immunokemi-og immunoblot-undersøgelser ikke entydige, hvilket indikerede unormalt / reduceret karrus, karrus, sarcoglycan, karrus-dystroglycan og dystrofinsignal kun i nekrotiske fibre (gentagen immunokemi resulterede normalt, også for laminin); immunoblots for dystrophin og calpain afslørede normale signaler. Imidlertid er calpain-3 immunoblot kendt for ikke at være helt følsom for LGMD2A diagnose, da 20-30% af tilfældene viser en normal mængde protein (1, 16-18). På grund af den kliniske præsentation (hyperCKmia, kalvehypertrofi/pseudo-hypertrofi) blev dystrofinopatier først udelukket. Desuden afslørede analyse af DMD (Hsp21), fkrp (19k13.32) og DYSF (2p13.2) gener ikke patogene mutationer. Patient kom til vores observation i en alder af 30, præsentere aksial og bælte involvering både i øvre og nedre lemmer, med betydelig vraltende gangart og vingede scapulae. I underekstremiteterne var svaghed fremtrædende i kvadriceps og glutei muskler, som var signifikant ipo-atrofiske sammen med beviset for “tilsyneladende hypertrofisk” (pseudo-hypertrofi) kalvemuskler (Figur 1). Kramper, myalgi og krusning blev ikke observeret. Komplet pneumologisk (spirometri og polysomnografi) og kardiologisk (ekkografi, 24-h EKG Holter-overvågning, hjerte-MR, komplet kardiologisk fysisk inspektion med målrettet medicinsk historie og kardiovaskulær refleksanalyse) undersøgelser afslørede ingen signifikant abnormitet. Vi udelukkede også syremaltasemangel (normale aktivitetsanalyser i lymfocytter/leukocytter). Muskelmagnetisk resonansafbildning (MRI) viste en fremtrædende involvering af skulderbælte, paravertebrale muskler, bageste rummuskler i låret (med relativ sparing af sartorius-og gracilis-musklerne) og af benet (især gastrocnemius medialis og soleus) (figur 2). Dette mønster af involvering er allerede rapporteret i litteraturen vedrørende MR-billede i calpainopati (19, 20). Derudover viste MR-undersøgelsen, at kalvemuskler effektivt var atrofiske og karakteriseret ved fedtinfiltration, hvilket forårsagede en muskuløs “forstørrelse.”Den kliniske præsentation, begyndelsesalderen, progressionshastigheden, fordelingen af muskelsvaghed og MR-fundene i proband var fuldt ud i overensstemmelse med en diagnose af LGMD2A, der i vid udstrækning er beskrevet i litteraturen (4, 21, 22).
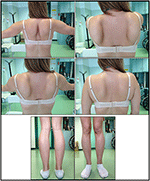
Figur 1. Proband kliniske træk: vingede scapulae og kombination af låratrofi og kalv pseudo-hypertrofi.
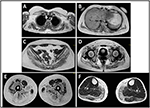
figur 2. Proband muskel MR: involvering af øvre lemmerbælte og paravertebrale muskler (A,B), involvering af nedre lemmerbælte og bageste rum i låret (C–E, for det meste glutei) med relativ sparing af sartorius og gracilis og involvering af det bageste rum i benet (F, for det meste gastrocnemius medialis/soleus).
evalueringen af familiemedlemmerne i proband afslørede ikke nogen historie med neuromuskulær sygdom og intet bevis for sammenhæng mellem forældre. Imidlertid præsenterede moderen til proband i en alder af 55 2 synkopale episoder, for hvilke hun blev diagnosticeret med idiopatisk bradykardi (op til 40 bpm om natten). Omfattende kardiologiske undersøgelser på moderen til proband diagnosticerede en symptomatisk atrioventrikulær (AV) blok efter forekomsten af flere syncopale episoder/lipothymi. Analysen viste tilstedeværelsen af en grundlæggende AV-blok i første grad med flere perioder med svær bradykardi, for det meste natlig og ofte symptomatisk, forbundet med nogle faser af anden grad AV-blok, både type 1 og 2, og nogle faser af natlig komplet AV-dissociation. Ekkokardiografi og ergometrisk test afslørede ikke signifikante ændringer, undtagen for patentet foramen ovale. Moderen præsenterede ikke noget andet klinisk symptom, prædisponerende tilstand eller risikofaktor. I betragtning af hendes kliniske billede blev probandens mor implanteret med PMK. Derudover afslørede familiehistorien, at bedstemor og oldefar til proband også blev implanteret med PMK i en alder af henholdsvis 55 og 30 år. Desuden døde bror til bedstemor til proband ved 51 år for alvorlig dilateret kardiomyopati. Kardiologiske undersøgelser blev også udført på Faderen og moderens onkel af Probanden, som resulterede fuldstændig upåvirket.
denne undersøgelse blev godkendt af etikudvalget for Santa Lucia Foundation og blev udført i henhold til Helsinki-erklæringen. Alle deltagere gav underskrevet informeret samtykke til genetisk analyse, og i den henseende gav de også samtykke til offentliggørelse af denne sagsrapport.
laboratorieundersøgelser og diagnostiske Tests
genomisk DNA blev ekstraheret fra perifert blod (400 liter) ved hjælp af Magpuriks blod-DNA-Ekstraktionssæt og Magpuriks automatisk Ekstraktionssystem (Resnova) i henhold til producentens anvisninger. Prøver blev sekventeret ved hjælp af Ion – PGM-System og Ionforstærket tilpasset Panelhøj specificitet (Thermo Fisher Scientific). Panelets størrelse var 129.13 Kb, som forventes at screene ~99,72% af det samlede panel med en minimumsdækning på 20 gange. panelet omfattede 18 gener, som blev udvalgt ved hjælp af videnskabelig litteratur, Generevisninger (www.ncbi.nlm.nih.gov/books/NBK1116/) og hyppighed af patogene varianter i den generelle befolkning. En detaljeret beskrivelse af NGS-panelet er opsummeret i tabel 1.
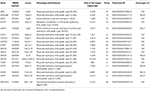
tabel 1. Tilpasset NGS panel anvendes til LGMD diagnose.
Biblioteksbyggeri blev udført af Ion Amplise kits 2.0. Ca.10 ng/prisl af start-DNA blev anvendt til multipleks PCR-reaktioner. Efter hinanden blev der udført to oprensningstrin for at fjerne uønskede forurenende stoffer, og en endelig PCR blev udført. Skabelon forstærkning og berigelse trin blev udført af Ion PGM hi-K OT2 kit-400, Ion OneTouch 2 System og Ion OneTouch ES (Thermo Fisher Scientific). Prøver blev behandlet af Ion PGM Hi-K sekventering Kit (400 bp, Thermo Fisher Scientific) og køre på Ion 316 Chip v2 (850 strømme kræves) og Ion PGM Sekvensator (Thermo Fisher Scientific). Resultaterne blev analyseret ved hjælp af Ion Reporter 4.6 (Thermo Fisher Scientific) og integreret Genomfremviser (IGV). Fortolkningen af genetiske varianter blev udført af Human Genmutationsdatabase (HGMD), Leiden Open Variation Database (LOVD), ClinVar og eks. Den funktionelle effekt af de detekterede varianter blev evalueret af bioinformatiske forudsigelsesværktøjer, herunder Mutationstaster, Varsome, SIFT, PolyPhen 2, SMART, Human Splejsning Finder (HSF). Direkte sekventering (BigDye Terminator v3.1, Bigdyeks Terminator og ABI3130, Applied Biosystems) blev udført for at bekræfte genetiske varianter og for at sekvensere genomiske kodende regioner med en dækning <20 gange (LMNA, Chr1:156106052-156106076; DYSF, Chr2:71753352-71753502, Chr2:71776385-71776640; SGCB, Chr4:52904225-52904560; Sgca, chr17:48243242-48243570).
resultater
NGS-analysen af proband afslørede 3 heterosygøse varianter (supplerende Figur 1). To varianter blev lokaliseret i CAPN3, nemlig NM_000070.2 (CAPN3): c.550dela (s.Thr184Argfs) og c.1813g>C (s.Val605Leu) i henholdsvis eksonerne 4 og 16. Den første er en enkelt nukleotidsletion og er den mest almindelige (75% af tilfældene) patogene variant i europæiske lande (1). Som forventet klassificerede den bioinformatiske analyse c.550dela (rs80338800) som en tab af funktionsvariant (s.Thr184Argfs), hvilket forårsagede en frameshift af den åbne læseramme. De forudsigelige værktøjer (Mutationstaster, HSF, Varsome, PolyPhen 2, SIFT) beskrev varianten som skadelig. Derudover afslørede SMART, at det ændrede proteinprodukt mangler calpain 3-domænet og de tre “EF-hænder” – motiver. Det første domæne er involveret i Calpain ‘ s signalveje, mens EF-hænderne er afgørende for Ca++-afhængig aktivering af proteinet (4).
vedrørende c.1813g > C, det er en ny missense-variant (S.Val605Leu), som er blevet forudsagt at være skadelig (ved Mutationstaster, HSF, Varsome, polyfen 2, Sigt) på grund af en potentiel ændring af splejsning. Denne variant er ikke kommenteret i litteraturen eller blandt online databaser, og den er ikke fundet hos 200 kontrolpersoner. Desværre gav analysen af SMART tool ikke signifikante resultater, da varianten er placeret inden for et ukarakteriseret domæne. Segregeringsanalysen af CAPN3 viste, at moderen og moderens onkel begge var heterosygøse for c.550delA, mens faderen var bærer af c.1813g>C.
derudover afslørede NGS-analyse på proband tilstedeværelsen af en ny variant i LMNA, nemlig NM_170707 (LMNA): c.550C>T (s.Gln184*). Den ovennævnte variant er forudsagt at være en null-variant, er endnu ikke beskrevet i litteratur eller blandt online databaser, og den er ikke fundet hos 200 kontrolpersoner. Prædiktive værktøjer (Mutationstaster, HSF, Varsome, PolyPhen 2, SIFT) beskrev en patogen virkning af denne variant på proteinproduktet. SMART tool rapporterede, at det ændrede lmna-protein mangler filamentdomænet, hvilket er vigtigt for at opretholde dets struktur og funktion. Segregeringsanalysen fremhævede tilstedeværelsen af c.550C>t-variant kun i moderen, hvilket tyder på en mulig sammenhæng med hendes hjerte symptomatologi og hendes familiehistorie af sygdom.
i henhold til kriterierne fastlagt af American College of Medical Genetics (ACMG) standarder og retningslinjer (23), c.1813g> C (CAPN3) kan beskrives som en sandsynlig patogen variant i betragtning af at den er placeret i et kritisk domæne uden godartet variation (PM1); det er fraværende i eks -, GnomAD-og 1000-genomdatabaser (PM2); i betragtning af recessiv arvsmodel er den blevet påvist i trans med en patogen variant (PM3); flere linjer med beregningsbevis understøtter en skadelig virkning på genet eller genproduktet (PP3). Med hensyn til den kliniske klassificering af C.550C>T (LMNA) af ACMG kan den betegnes som en patogen variant, da det er en null-variant, der forårsager tab af funktion (S.Gln184*) i LMNA (PVS1); det er fraværende i eks -, GnomAD-og 1000-genomdatabaser (PM2); det er placeret i et kritisk domæne uden godartet variation (PM1); flere linjer med beregningsbevis understøtter en skadelig virkning på genet eller genproduktet (PP3).
konklusioner
denne sagsrapport præsenterede en patient påvirket af LGMD, som viste sig at være bærer af mutationer ikke kun i CAPN3 (c.550dela og c.1813g>C) men også i LMNA (c.550C > T). Disse resultater forklarer probandens neuromuskulære fænotype på grund af CAPN3-mutationer og fremhæver en potentiel risiko for hjerte-kar-sygdomme på grund af tilstedeværelsen af varianten i LMNA og hendes positive familiehistorie. I betragtning af disse resultater blev familiemedlemmerne udsat for segregeringsanalysen. Især resulterede moderen i at være bærer af henholdsvis CAPN3_c.550dela og LMNA_c.550C>T. Moderen kan betragtes som en sund bærer for capn3_c.550dela patogen mutation forbundet med calpainopati. Tilstedeværelsen af en ny variant i LMNA kan dog forklare hendes kardiovaskulære patologi (bradykardi og syncopale episoder). Fraværet af neuromuskulær symptomatologi hos moderen og det særegne kliniske billede af proband udelukket en mulig forening af lmna_c.550C> T med neuromuskulær fænotype, især vedrørende Emery-Dreifuss muskeldystrofi (EMD). Faktisk påvirker EMD specifikt vastus lateralis og biceps brachii muskler, som er relativt skånet i LGM2A (24, 25).
morbroren var kun heterosygøs for CAPN3_c.550dela og viste som forventet ingen neuromuskulære eller kardiovaskulære problemer. Faderen resulterede i at bære romanen CAPN3_c.1813g>C og dermed var han upåvirket. Samlet set bekræftede segregationsanalyse arven af de tre mutationer af Probanden fra hendes slægtninge og fremhævede en fortrolighed for kardiomyopati, som ikke kan overses (figur 3).
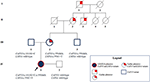
figur 3. Stamtavle, der viser den positive fortrolighed for hjertefænotype arvet af moderens afstamning og transmission af capn3-og LMNA-varianterne i hele familiemedlemmerne.
alt i alt rejser disse data nogle vigtige overvejelser. Først, NGS-panel i denne patient var kritisk for at nå den molekylære diagnose af calpainopati, påvisning af en kendt mutation og en anden ny variant forudsagt som patogen. For det andet tillod NGS-panelet identifikationen af den nye lmna_c.550C>t-variant i proband. Dette resultat var i overensstemmelse med den positive familiehistorie og med segregeringsdata, der forklarede hjerte-kar-sygdomme hos moderen og, endnu vigtigere, anbefalede mere specifik kardiologisk opfølgning i proband. Det er vigtigt at bemærke, at kardiologiske manifestationer forekom hos moderen senere i alderen (55 år), så vi kan antage, at Probanden stadig kunne være i en “asymptomatisk kardiologisk fase.”Imidlertid kan en variabel fænotypisk ekspression af LMNA-mutation i Probanden sammenlignet med moderen ikke udelukkes. Alle disse data understreger vigtigheden af en integreret tilgang mellem klinikere og genetikere for korrekt fortolkning af resultater, korrekt genetisk rådgivning og til sidst klinisk styring og opfølgning. Med hensyn til genetisk og familiær rådgivning blev NGS-analysen også udført på probandens partner for at estimere reproduktionsrisikoen for parret. Partneren var negativ for alle de 18 testede gener, hvilket betyder, at han har 1/650 restrisiko for at være en sund bærer for LGMD-årsagsmutationer, i betragtning af at testen er 84% følsom. Under hensyntagen til probandens genetiske profil og følsomheden af NGS-testen er den resterende risiko for parret at få et barn ramt af calpainopati 1/1300. På den anden side overføres lmna-patogene varianter i henhold til et autosomalt dominerende mønster, hvilket betyder, at Probanden har 50% sandsynlighed for at have heterosygøse børn. Imidlertid kan det kliniske billede af afkom ikke helt sikkert forudsiges, da LMNA_c.550C>T er en ny variant, og dens funktionelle indvirkning på fænotypen er stadig ukendt.
afslutningsvis fremhæver denne sagsrapport den kliniske anvendelighed af NGS-paneler til at give nøjagtig LGMD2A-diagnose og beskrive komplekse fænotyper og comorbiditeter, der stammer fra arv af forskellige mutationer i flere gener. Anvendelsen af NGS i den kliniske praksis bør dog altid kombineres med en præ – og postgenetisk rådgivning for at give en klar forklaring af resultaterne, de mulige konsekvenser for patienternes fænotype, tilbagefaldsrisikoen i familien samt for at forklare mulige uventede fund.
datatilgængelighed
ingen datasæt blev genereret eller analyseret til denne undersøgelse.
Etikerklæring
denne undersøgelse blev udført i overensstemmelse med anbefalingerne fra Santa Lucia Foundation ‘ s etiske udvalg med skriftligt informeret samtykke fra alle fag. Alle emner gav skriftligt informeret samtykke i overensstemmelse med Helsinki-erklæringen. Protokollen blev godkendt af etikudvalget for Santa Lucia Foundation.
Forfatterbidrag
RC, CSt, VC, GC, RG, GPa, SC, CP og JM bidrog til erhvervelse af data, analyse og fortolkning af data. CSt, RC, GC, RG, GM og EG har været involveret i udarbejdelsen af manuskriptet. GPR, CSA og SS har været involveret i erhvervelsen af kliniske data. RC, CST, VC, GC, RG, GPA, SC, CP, JM, GPR, GM, CSa, SS og EG har givet endelig godkendelse af den version, der skal offentliggøres.
Funding
dette arbejde er støttet af 5h2016 National Health Ministry 2.
interessekonflikt Erklæring
forfatterne erklærer, at forskningen blev udført i mangel af kommercielle eller økonomiske forhold, der kunne fortolkes som en potentiel interessekonflikt.
supplerende materiale
det supplerende materiale til denne artikel kan findes online på: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. Protein og genetisk diagnose af muskeldystrofi i lemmerbæltet type 2a: udbyttet og faldgruberne. Muskel Nerve. (2015) 52:163–73. doi: 10.1002 / mus.24682
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
2. Nigro V, Savarese M. genetisk grundlag for muskeldystrofier i lemmer: opdateringen fra 2014. Acta Myol. (2014) 33:1–12.
PubMed Abstrakt / Google Scholar
3. Richard i, Hogrel JY, Stockholm D, Payan CAM, Fougerousse F, Eymard B, et al. Naturhistorie af LGMD2A til afgrænsning af resultatmål i kliniske forsøg. Ann Clin Transl Neurol. (2016) 3:248–65. doi: 10.1002 / acn3.287
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
4. Angelini C, Fanin M. Calpainopathy. I: Adam MP, Ardinger HH, Pagon RA redaktører. Generevurderinger. Aalborg Universitet (2017) 1-30.
PubMed Abstrakt / Google Scholar
5. Kyriakides T, Angelini C, Schaefer J, Sacconi S, Siciliano G, Vilchesjj, et al. Efns retningslinjer for den diagnostiske tilgang til pauci – eller asymptomatisk hyperckæmi. Eur J Neurol. (2010) 17:767–73. doi: 10.1111 / j. 1468-1331.2010. 03012.
CrossRef Fuld tekst / Google Scholar
6. Mori-Yoshimura M, Sega K, Minami N, Oya Y, Komaki H, Nonaka i, et al. Kardiopulmonal dysfunktion hos patienter med muskeldystrofi i lemmer-bælte 2A. muskel Nerve. (2017) 55:465–9. doi: 10.1002 / mus.25369
CrossRef Fuld Tekst | Google Scholar
7. Okere a, Reddy SS, Gupta s, Shinnar M. A kardiomyopati hos en patient med muskeldystrofi af lemmer type 2a. Circ hjertesvigt. (2013) 6:e12–13. doi: 10.1161 / CIRCHEARTFAILURE.112.971424
CrossRef Fuld Tekst | Google Scholar
8. Kramerova I, Ermolova N, Eskin A, Hevener A, Hehenberger O, Armando AM, et al. Manglende regulering af transkription af gener, der er nødvendige for muskeltilpasning, ligger til grund for muskeldystrofi i lemmer 2A (calpainopati). Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093 / hmg / dd086
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
9. Thompson R, Straub V. lemmer bælte muskeldystrofier-internationale samarbejder for translationel forskning. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038 / nrneurol.2016.35
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
10. Strafella C, Caputo V, Galota MR, Marella G, Mauriello S, et al. Anvendelse af præcisionsmedicin i neurodegenerative sygdomme. Front Neurol. (2018) 9:701. doi: 10.3389 / fneur.2018.00701
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
11. Cascella R, Strafella C, Caputo V, Errichiello V, Milano F, et al. Mod anvendelse af præcisionsmedicin i aldersrelateret makuladegeneration. Prog Retin Eye Res. (2017) 63:132-46. doi: 10.1016 / j. preteyeres.2017.11.004
PubMed Abstrakt | CrossRef Fuld Tekst | Google Scholar
12. Cascella R, Strafella C, Longo G, Manso L, De Felici C, et al. Vurdering af individuel risiko for AMD med genetisk rådgivning, familiehistorie og genetisk testning. Øjne. (2017) 32:446–50. doi: 10.1038 / øje.2017.192
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
13. Adams DR, Eng CM. Næste generations sekventering til diagnosticering af mistænkte genetiske lidelser. N Engl J Med. (2019) 379:1353–62. doi: 10.1056 / NEJMc1814955
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
14. Angelini C. neuromuskulær sygdom. Diagnose og opdagelse i muskeldystrofi i lemmer. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038 / nrneurol.2015.230
CrossRef Fuld Tekst | Google Scholar
15. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SV, et al. Anvendelse af heleksomsekventering til diagnose af muskeldystrofi i lemmer: resultater og erfaringer. JAMA Neurol. (2015) 72:1424–32. doi: 10.1001 / jamaneurol.2015.2274
PubMed Abstrakt | CrossRef Fuld Tekst | Google Scholar
16. Milic A, Daniele N, Lochm Larsller H, Mora M, Comi GP, Moggio M, et al. En tredjedel af LGMD2A biopsier har normal calpain 3 proteolytisk aktivitet som bestemt ved en in vitro-analyse. Neuromusc Disord. (2007) 17:148–56. doi: 10.1016 / j. nmd.2006.11.001
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
17. F, F, F, et al. Tidlig debut calpainopati med normalt ikke-funktionelt calpain 3-niveau. Dev Med Child Neurol. (2006) 48:304–6. doi: 10.1017/S001216220600065
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
18. Talim B, Ognibene a, Mattioli E, Richard i, Anderson LV, Merlini L. Normal calpain-ekspression i genetisk bekræftet muskeldystrofi af lemmer type 2a. neurologi. (2001) 56:692–3. doi: 10.1212 / VNL.56.5.692-a
CrossRef Fuld tekst / Google Scholar
19. Llauger J, Gallardo E, Illa I. muskel MR i muskeldystrofier. Acta Myol. (2015) 34:95–108.
PubMed Abstrakt / Google Scholar
20. Mercuri E, Bushby K, Ricci E, Birchall DR, Pane M, Kinali M, et al. Muskel-MR-fund hos patienter med muskeldystrofi i lemmer med calpain 3-mangel (LGMD2A) og tidlige kontrakturer. Neuromusc Disord. (2005) 15:164–71. doi: 10.1016 / j. nmd.2004.10.008
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
21. Magri F, Nigro V, Angelini C, Mongini T, Mora M, Moroni I, et al. Det italienske register over muskeldystrofi i lemmer: relativ frekvens, kliniske træk og differentiel diagnose. Muskel Nerve. (2017) 55:55–68. doi: 10.1002 / mus.25192
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
22. Chae J, Minami N, Jin Y, Nakagava M, Murayama K, Igarashi F, et al. Calpain 3 genmutationer: genetiske og klinisk-patologiske fund i muskeldystrofi i lemmer. Neuromusc Disord. (2001) 11:547–55. doi: 10.1016 / S0960-8966(01)00197-3
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
23. Richards S, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standarder og retningslinjer for fortolkning af sekvensvarianter: en fælles konsensusanbefaling fra American College of Medical Genetics and Genomics og Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038 / gim.2015.30
PubMed Abstrakt | CrossRef Fuld Tekst | Google Scholar
24. Bonne G, Mercuri E, Muchir A, Urtisberea A, B Larscane HM, Recan D, et al. Klinisk og molekylærgenetisk spektrum af autosomal dominerende Emery-Dreifuss muskeldystrofi på grund af mutationer af lamin A/C-genet. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170::støtte-ANA6>3.0.CO; 2-J
PubMed abstrakt / CrossRef fuldtekst / Google Scholar
25. Hong JS, ki CS, Kim JV, Suh YL, Kim JS, Baek KK, et al. Hjertearytmier, kardiomyopati og muskeldystrofi hos patienter med Emery-Dreifuss muskeldystrofi og muskeldystrofi i lemmer type 1B. J Korean Med Sci. (2005) 20:283–90. doi: 10.3346 / jkms.2005.20.2.283
CrossRef Fuld Tekst | Google Scholar