- introduktion
- metoder
- filtrering af ribosomale Profileringsdata
- samling af virtuelle genomer
- bestemmelse af Ribo-sek Read-Mapped Region på et kryds (RMRJ) af circrna ‘ er
- træning af modellen og klassificering af RMRJs
- forudsigelse af oversatte peptider af RMRJs
- resultater og diskussion
- oversatte circRNAs hos mennesker og A. thaliana
- funktionel berigelse af humane og A. thaliana circrna ‘ er med Kodningspotentiale
- Nøjagtighedstest for CircCode
- indflydelse af Ribosekvensdybde
- sammenligning af CircCode med andre værktøjer
- konklusioner
- tilgængelighed og krav
- Erklæring om tilgængelighed af Data
- Forfatterbidrag
- finansiering
- interessekonflikt
- supplerende materiale
introduktion
cirkulære RNA ‘er (circrna’ er) er en speciel type ikke-kodende RNA-molekyle, der er blevet et varmt forskningsemne inden for RNA og får stor opmærksomhed (Chen og Yang, 2015). Sammenlignet med traditionelle lineære RNA’ er (indeholdende 5′ og 3 ‘ ender) har circRNA-molekyler normalt en lukket cirkulær struktur; hvilket gør dem mere stabile og mindre tilbøjelige til nedbrydning (Vicens og Vesthof, 2014). Selvom eksistensen af circrna ‘ er har været kendt i nogen tid, blev disse molekyler betragtet som et biprodukt af RNA-splejsning. Men med udviklingen af high-throughput sekventering og bioinformatik teknologier, circrna ‘ er er blevet bredt anerkendt i dyr og planter (Chen og Yang, 2015). Nylige undersøgelser har også vist, at et stort antal circrna ‘ er kan oversættes til små peptider i celler (Pamudurti et al., 2017) og har nøgleroller på trods af deres undertiden lave udtryksniveau (Hsu og Benfey, 2018; Yang et al., 2018). Selv om et stigende antal circrna ‘ er identificeres, er deres funktioner i planter og dyr generelt stadig at blive undersøgt. Ud over deres funktioner som miRNA-lokkefugle har circrna ‘ er et vigtigt translationspotentiale, men der er ingen værktøjer til rådighed til specifikt at forudsige disse molekylers translationelle evner (Jakobi og Dieterich, 2019).
der findes flere værktøjer til forudsigelse og identifikation af circrna ‘ er, såsom CIRI (Gao et al., 2015), Cirkusforløber (Dong et al., 2019), CircPro (Meng et al., 2017), og circtools (Jakobi et al., 2018). Blandt dem kan CircPro afsløre oversatte circrna ‘er ved at beregne en oversættelsespotential score for circrna’ er baseret på CPC (Kong et al., 2007), som er et værktøj til at identificere den åbne læseramme (ORF) i en given rækkefølge. Men fordi nogle circrna ‘ er ikke bruger startkodonet under oversættelse (Ingolia et al., 2011; Slavoff et al., 2013; Kearse, 2017; Spealman et al., 2018), der anvender CPC, kan filtrere nogle virkelig oversatte circrna ‘ er ud. I denne undersøgelse brugte vi BASiNET (Ito et al., 2018), som er en RNA-klassifikator baseret på maskinindlæringsmetoderne (random forest og J48 model). Det transformerer oprindeligt de givne kodende RNA ‘er (positive data) og ikke-kodende RNA’ er (negative data) og repræsenterer dem som komplekse netværk; det udtrækker derefter de topologiske mål for disse netværk og konstruerer en funktionsvektor til at træne den model, der bruges til at klassificere kodningskapaciteten for circrna ‘ er. Med denne metode undgås fejlagtig filtrering af oversatte circrna ‘ er, der ikke er initieret af AUG. Derudover er Ribo-sekv-teknologi, der er baseret på sekventering med høj kapacitet til at overvåge RPF ‘ er (ribosomale beskyttede fragmenter) af udskrifter (Guttman et al., 2013; Brar og Vaissman, 2015), kan bruges til at bestemme placeringen af circrna ‘ er, der oversættes (Michel and Baranov, 2013). For at identificere circRNAs kodningsevne udviklede vi værktøjet CircCode, som involverer en Python 3–baseret ramme og anvendte CircCode til at undersøge oversættelsespotentialet for circRNAs fra mennesker og Arabidopsis thaliana. Vores arbejde giver en rig ressource til yderligere undersøgelse af funktionerne i circrna ‘ er med kodningskapacitet.
metoder
CircCode blev skrevet i Python 3 programmeringssprog; det bruger Trimmomatic (Bolger et al., 2014), Butterfly (Langmead og Salberg, 2012) og STAR (Dobin et al., 2013) for at filtrere rå Ribo-sek læser og kortlægge disse filtrerede læser til genomet. CircCode identificerer derefter Ribo-sek read-mapped regioner i circrna ‘ er, der indeholder kryds. Derefter sorteres de kandidatkortede sekvenser i circrna ‘erne baseret på klassifikatorer (J48-model) i kodende RNA’ er og ikke-kodende RNA ‘ er efter BASiNET. Endelig identificeres korte peptider produceret ved translation som potentielle kodende regioner af circrna ‘ er. Hele processen med CircCode består af fem trin (Figur 1).
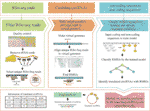
Figur 1 arbejdsgangen for CircCode. Det øverste lag repræsenterer den inputfil, der kræves for hvert trin i CircCode. Mellemlaget er opdelt i tre dele, og hver del repræsenterer et andet driftsstadium. Fra venstre mod højre repræsenterer den første del filtreringen af Ribosekv-dataene; kvalitetskontrollen udføres af Trimmomatic, og rRNA-læsningerne fjernes med Butterfly. Den anden del repræsenterer de trin, der bruges til at producere det virtuelle genom og justere de filtrerede læsninger til det virtuelle genom med stjerne. Den sidste del repræsenterer identifikationen af oversatte circrna ‘ er ved maskinindlæring. Bundlaget repræsenterer det sidste trin, der blev brugt til at forudsige peptiderne oversat fra circrna ‘erne og de endelige outputresultater, herunder information om oversatte circrna’ er og deres oversættelsesprodukter.
filtrering af ribosomale Profileringsdata
først fjernes fragmenter og adaptere af lav kvalitet i Ribosekv-aflæsningerne af Trimmomatic med standardparametrene for at opnå rene Ribosekv-aflæsninger. For det andet kortlægges disse rene Ribosekv-læsninger til et rRNA-bibliotek for at fjerne læsninger afledt af rRNA ved hjælp af Butterfly. Fordi læselængderne på Ribosekv er relativt korte (generelt mindre end 50 bp), er det muligt for en læsning at matche flere regioner. I dette tilfælde er det vanskeligt at bestemme, hvilken region en bestemt læsning svarer til. For at undgå dette kortlægges de rene Ribosekv-læsninger til genomet af en art af interesse, og læsninger, der ikke er perfekt tilpasset genomet, betragtes som de endelige unikke Ribosekv-læsninger.
samling af virtuelle genomer
Circrna ‘ er vises normalt som ringformede molekyler i eukaryoter, og de kan identificeres ud fra deres splejsningskryds. Imidlertid er sekvenserne af circrna ‘ er i fasta-filen ofte i lineær form. I teorien indikerer resultatet, at krydset er mellem det 5′ terminale nukleotid og det 3’ terminale nukleotid, skønt krydset og sekvensen nær krydset ikke kan ses direkte, hvorved Ribosekvens læses til circRNA-sekvenser, inklusive kryds, på en ligetil måde.
CircCode forbinder sekvensen af hvert circRNA i tandem, således at krydset for hver er midt i den nybyggede sekvens. Vi adskilt også hver serieenhed med 100 N nukleotider for at undgå forvirring ved sekvensjusteringstrinnet (længden af hver RPF er mindre end 50 bp). Endelig opnåede vi et virtuelt genom, der kun bestod af kandidatcirkrna ‘ er i tandem adskilt af 100 Ns. Fordi CircCode kun fokuserer på justering mellem Ribosekvenslæsninger og circRNA-sekvenser, kan vi undersøge kodningspotentialet for circrna ‘er ved at kortlægge Ribosekvenslæsningerne til dette virtuelle genom, hvilket kan spare en stor mængde beregningstid (det virtuelle genom er meget mindre end hele genomet) og øge nøjagtigheden (ved at undgå interferens mellem opstrøms og nedstrøms sekvenssammenligninger af circrna’ erne).
bestemmelse af Ribo-sek Read-Mapped Region på et kryds (RMRJ) af circrna ‘ er
de endelige unikke Ribo-sek læser er kortlagt til et tidligere oprettet virtuelt genom ved hjælp af stjerne. Fordi hver tandem circRNA-enhed blev adskilt af 100 N baser, før de producerede det virtuelle genom, blev den største intronlængde indstillet til ikke at overstige 10 baser med parameteren “–alignintronmaks 10.”Denne parameter eliminerer enhver interaktion mellem forskellige cirkrna’ er i sekvensjusteringen. I det andet trin i virtuel genomproduktion gemmer CircCode positionelle krydsoplysninger for hvert circRNA i det virtuelle genom. Hvis Ribo-sek read-mapped region i det virtuelle genom inkluderer krydset mellem circRNA, og antallet af kortlagte Ribo-sek læser på krydset (NMJ) er større end 3, kan Ribo-sek læser-kortlagt region ved krydset mellem circrna ‘erne betragtes som en RMRJ, som afslører et groft oversat segment af circrna’ er nær krydsningsstedet.
træning af modellen og klassificering af RMRJs
selvom RMRJs kan udgøre et stærkt bevis på oversættelse, er der stadig nogle mangler i denne metode. Fordi længden af læsningerne på ribosomalkortet er kort, kan en læsning sammenlignes med den forkerte position. Derfor er det ikke overbevisende blot at betragte den region, der er omfattet af Ribosekv, som den oversatte region. Til dette formål anvendes maskinindlæringsmetoden til at identificere rmrj ‘ s kodningsevne. For det første udtrækker CircCode kodning af RNA ‘er (positive data) og ikke-kodende RNA’ er (negative data) fra en art af interesse og bruger dem til modeluddannelse ved hjælp af forskellen i funktionsvektorer mellem kodning og ikke-kodende RNA ‘ er. CircCode bruger derefter den uddannede model til at klassificere RMRJs opnået i det foregående trin ved BASiNET. Hvis rmrj for et circRNA genkendes som kodende RNA, kan dette circRNA identificeres som et oversat circRNA.
forudsigelse af oversatte peptider af RMRJs
da ekspression af cirkrna ‘er i organismer er lav, viser Ribosekv-data ikke den nøjagtige 3-NT-periodicitet klart i tilfælde af færre RPF’ er. Derfor er det vanskeligt at bestemme det nøjagtige oversættelsesstartsted for et oversat circRNA. På grund af tilstedeværelsen af et stopkodon i nogle Rmrj ‘ er, og fordi startkodonet er vanskeligt at bestemme, er metoden til at finde en ORF baseret på et startkodon og et stopkodon ikke mulig.
for at bestemme de sande oversættelsesregioner for disse circrna ‘ er og generere det endelige oversættelsesprodukt, FragGeneScan (Rho et al., 2010), som kan forudsige proteinkodende regioner i fragmenterede gener og gener med frameshifts, bruges til at bestemme de oversatte peptider produceret af circrna ‘ er.
for at undgå den besværlige køreproces kan alle modeller kaldes af et shell-script; brugeren kan blot udfylde den givne konfigurationsfil og indtaste den i script, og hele processen til forudsigelse af de oversatte circrna ‘ er køres derefter. Derudover kan CircCode køres separat, trin for trin, således at brugeren kan justere parametrene midt i proceduren og se resultaterne af hvert trin efter ønske.
resultater og diskussion
efter test på flere computere blev CircCode fundet at køre med succes med de nødvendige afhængigheder installeret. For at teste udførelsen af CircCode brugte vi data for mennesker og A. thaliana til at forudsige circrna ‘ er med oversættelsespotentiale. Resultaterne blev sammenlignet med circrna ‘ er, der er verificeret eksperimentelt som bekræftelse. Derefter testede vi den falske opdagelsesrate (FDR) værdi af CircCode yderligere. Vi brugte GenRGenS (Ponty et al., 2006) at generere et datasæt til test baseret på kendte oversatte circrna ‘ er og bekræftede, at FDR-værdien var inden for et acceptabelt interval og på et lavt niveau. Endelig evaluerede vi effekten af forskellige sekventeringsdybder af Ribosekv-data på Cirkodeforudsigelser og sammenlignede CircCode med andre programmer.
oversatte circRNAs hos mennesker og A. thaliana
for at anvende CircCode-værktøjet på rigtige data hentede vi først filerne inklusive det humane referencegenom GRCh38, genom-annotation og human rRNA fra Ensembl. For A. thaliana blev referencegenomerne (TAIR10), genom-annotationsfiler og tilsvarende rRNA-sekvenser alle hentet fra Ensembl-planter. Data for mennesker og A. thaliana blev hentet fra rpfdb (tiltrædelsesnumre: GSE96643, GSE81295, GSE88794) (Hsu et al., 2016; et al., 2017), og alle kandidatcirkrnas fra human og A. thaliana blev hentet fra CIRCPedia v2 (Dong et al., 2018) og PlantcircBase, henholdsvis (Chu et al., 2017). I sidste ende identificerede vi 3.610 oversatte circrna ‘er fra mennesker og 1.569 oversatte circrna’ er fra A. thaliana ved hjælp af CircCode (supplerende Data 1).
funktionel berigelse af humane og A. thaliana circrna ‘ er med Kodningspotentiale
brug af CircCode-resultaterne for human og A. thaliana, onlineværktøjet KOBAS 3.0 (vu et al., 2006) blev ansat til at kommentere disse oversatte circrna ‘ er baseret på deres forældregener. Desuden udførte vi Go (Gen ontologi) funktionel analyse og KEGG (Kyoto Encyclopedia of Genes and Genomes) berigelsesanalyse for disse oversatte circrna ‘ er ved hjælp af R-pakken clusterProfiler (Yu et al., 2012).
KEGG-resultaterne viste, at de humane circrna ‘ er blev beriget i proteinbehandling i den endoplasmatiske retikulumvej, kulstofmetabolismevej og RNA-transportvej. GO-analyse indikerede deltagelse af humane oversatte circrna ‘ er i reguleringen af molekylebinding, ATPase-aktivitet og andre RNA-splejsningsrelaterede biologiske processer. Derudover er de oversatte circrna ‘ er fra A. thaliana beriget i veje relateret til stressmodstand, hvilket antyder, at de spiller vigtige roller i denne proces (supplerende Data 2).
Nøjagtighedstest for CircCode
for at undersøge nøjagtigheden af CircCode blev testsekvenser genereret af GenRGenS, som bruger den skjulte Markov-model til at producere sekvenser, der har de samme sekvensegenskaber (såsom frekvenserne af forskellige nukleotider, forskellige kodoner og forskellige nukleotider i starten af sekvensen) anvendt.
til denne undersøgelse brugte vi tidligere offentliggjorte humane oversatte circRNAs (Yang et al., 2017) som input til GenRGenS og genererede 10.000 sekvenser for at teste CircCode. Vi gentog testen 10 gange, og i gennemsnit blev 27 oversatte circrna ‘ er forudsagt hver gang. FDR-værdien blev beregnet til at være 0,0027, hvilket er meget mindre end 0,05, hvilket indikerer, at de forudsagte resultater er troværdige.
derudover sammenlignede vi de oversatte circrna ‘ er fra mennesker som identificeret ved CircCode med verificerede polysome-associerede circRNA-data (Yang et al., 2017). Blandt dem blev 60% af cirkrna ‘ erne identificeret ved hjælp af CircCode (supplerende Data 3).
indflydelse af Ribosekvensdybde
for at undersøge virkningen af sekventeringsdybden af Ribosekvensdata på Cirkodeidentifikationsresultaterne testede vi først effekten af sekventeringsdybde på antallet af oversatte cirkrna ‘ er (Figur 2a). Når sekventeringsdybden var lav, var det forudsagte antal oversatte cirkrna ‘er lavt, og antallet af oversatte cirkrna’ er steg med stigende sekventeringsdybde. Antallet af oversatte circrna ‘ er blev stabilt, da sekventeringsdybden nåede ikke mindre end 10 liters lineær udskriftsdækning.

figur 2 (a) effekt af ribosekvensdybde på det forudsagte antal oversatte cirkrna ‘ er. (B) virkningen af kryds læse nummer (JRN) på CircCode følsomhed på forskellige sekventeringsdybder.
for det andet blev indflydelsen af NMJ på følsomhed ved forskellige sekventeringsdybder også vurderet (figur 2b). Resultaterne viste, at NMJ havde mindre indflydelse på følsomheden, da sekventeringsdybden steg. CircCode havde også højere følsomhed ved brug af Ribosekv-data med højere sekventeringsdybde.
sammenligning af CircCode med andre værktøjer
for at sammenligne CircCode med andre værktøjer, såsom CircPro, det samme sæt Ribosekv-data (SRR3495999) fra A. thaliana blev brugt til at identificere oversatte circrna ‘ er ved hjælp af seks processorer med 16 gigabyte RAM. CircPro identificerede 44 oversatte circrna ‘er på 13 min, mens CircCode identificerede 76 oversatte circrna’ er på 20 min. CircCode er således mere følsom end CircPro på samme computerniveau, men det tager mere tid. CircPro er kortfattet og mindre tidskrævende end CircCode, men CircCode kan identificere flere circrna ‘ er med kodningsevne end CircPro.
konklusioner
Circrna ‘er spiller en vigtig rolle i biologi, og det er afgørende at nøjagtigt identificere circrna’ er med kodningsevne til efterfølgende forskning. Baseret på Python 3 udviklede vi CircCode, et brugervenligt kommandolinjeværktøj, der har høj følsomhed til at identificere oversatte circrna ‘ er fra Ribo-sek læser med høj nøjagtighed. CircCode udviser god præstation i både planter og dyr. Fremtidig arbejde vil tilføje nedstrøms karakteranalyse til CircCode ved at visualisere hvert trin i processen og optimere nøjagtigheden af forudsigelsen.
tilgængelighed og krav
CircCode er tilgængelig på https://github.com/PSSUN/CircCode; operativsystem(er): Python, programmeringssprog: Python 3 og R; andre krav: bedtools (version 2.20.0 eller nyere), Butterfly, STAR, Python 3 pakker (Biopython, Pandas, rpy2), R-pakker (BASiNET, Biostrings). Installationspakkerne til alle de nødvendige programmer er tilgængelige på CircCode-hjemmesiden. Brugere behøver ikke at hente dem individuelt. CircCode-hjemmesiden indeholder også detaljerede brugermanualer til reference. Værktøjet er frit tilgængeligt. Der er ingen begrænsninger for brug af nonacademics.
Erklæring om tilgængelighed af Data
alle relevante data findes i manuskriptet og dets understøttende informationsfiler.
Forfatterbidrag
konceptualisering: PS, GL. Databehandling: PS, GL. Formel analyse: PS, GL. Skrivning-originalt udkast: PS, GL. Skrivning-gennemgang og redigering: PS, GL.
finansiering
dette arbejde blev støttet af tilskud fra National Natural Science Foundation of China (grant nr. 31770333, 31370329 og 11631012), programmet for nye århundredes fremragende talenter på universitetet (NCET-12-0896) og de grundlæggende forskningsmidler til de centrale universiteter (nr. GK201403004). Finansieringsbureauerne havde ingen rolle i undersøgelsen, dens design, dataindsamling og analyse, beslutningen om at offentliggøre eller forberedelsen af manuskriptet. Finansiererne havde ingen rolle i studiedesign, dataindsamling og analyse, beslutning om at offentliggøre eller forberedelse af manuskriptet.
interessekonflikt
forfatterne erklærer, at forskningen blev udført i mangel af kommercielle eller økonomiske forhold, der kunne fortolkes som en potentiel interessekonflikt.
supplerende materiale
det supplerende materiale til denne artikel kan findes online på: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
supplerende Data 1 / sekvensen af det forudsagte oversatte circRNA og kort peptid.
supplerende Data 2 | GO berigelse og KEGG berigelse resultater for mennesker og Arabidopsis thaliana.
supplerende Data 3 | sammenligning af forudsagte oversatte circrna ‘er med validerede oversatte circrna’ er.
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: en fleksibel trimmer til illumina sekvensdata. Bioinformatik 30, 2114-2120. doi: 10.1093 / bioinformatik / btu170
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Brar, G. A., Viissman, J. S. (2015). Ribosomprofilering afslører hvad, hvornår, hvor og hvordan proteinsyntese. Nat. Pastor Mol. Celle Biol. 16, 651–664. doi: 10.1038 / nrm4069
PubMed abstrakt / CrossRef fuldtekst / Google Scholar
Chen, L.-L., Yang, L. (2015). Regulering af circRNA biogenese. RNA Biol. 12, 381–388. doi: 10.1080/15476286.2015.1020271
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
C., Mao, L., Ye, C., Et Al. (2017). PlantcircBase: en database for plante cirkulære RNA ‘ er. Mol. Plante 10, 1126-1128. doi: 10.1016 / j. molp.2017.03.003
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
det er en af de mest almindelige måder at gøre det på. (2013). Stjerne: ultrahurtig universal RNA-sekv aligner. Bioinformatik 29, 15-21. doi: 10.1093 / bioinformatik / bts635
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Dong, R., Ma, K.-K., Chen, L.-L., Yang, L. (2019). “Genom-dækkende annotation af cirkrna’ er og deres alternative back-splejsning/splejsning med Cirkuseksplorer Pipeline,” i Epitranscriptomics. EDS. N., Gupta, R. (Ny York, NY: Springer Ny York), 137-149. doi: 10.1007/978-1-4939-8808-2_10
CrossRef fuldtekst / Google Scholar
Dong, R., Ma, K.-K., Li, G.-V., Yang, L. (2018). CIRCpedia v2: en opdateret database til omfattende cirkulær RNA-annotation og ekspressionssammenligning. Genomics Proteomics Bioinf. 16, 226–233. doi: 10.1016 / j. gpb.2018.08.001
CrossRef Fuld Tekst / Google Scholar
Gao, Y., Vang, J., J., F. (2015). CIRI: en effektiv og upartisk algoritme til de novo cirkulær RNA-identifikation. Genom Biol. 16, 4. doi: 10.1186 / s13059-014-0571-3
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
(2013). Ribosomprofilering giver bevis for, at store ikke-kodende RNA ‘ er ikke koder for proteiner. Celle 154, 240-251. doi: 10.1016 / j.celle.2013.06.009
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
Hsu, P. Y., Benfey, P. N. (2018). Lille, men mægtig: funktionelle peptider kodet af små Orf ‘ er i planter. PROTEOMICS 18, 1700038. doi: 10.1002 / pmic.201700038
CrossRef Fuld tekst / Google Scholar
Hsu, P. Y., Calviello, L., Vu, H.-Y. L., Li, F.-V., Rothfels, C. J., Ohler, U., et al. (2016). Ribosomprofilering med superopløsning afslører uanmeldte oversættelsesbegivenheder i Arabidopsis. Proc. Natl. Acad. Sci. 113, E7126-E7135. doi: 10.1073 / pnas.1614788113
CrossRef Fuld Tekst / Google Scholar
Ingolia, N. T., Lareau, L. F., Viissman, J. S. (2011). Ribosomprofilering af embryonale stamceller fra mus afslører kompleksiteten og dynamikken i pattedyrsproteomer. Celle 147, 789-802. doi: 10.1016 / j.celle.2011.10.002
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Ito, E. A., Katahira, I., Vicente, F. F., da, R., Pereira, L. F. P., Lopes, F. M. (2018). BASiNET-biologiske Sekvensnetværk: et casestudie om kodning og ikke-kodende RNA-identifikation. Nukleinsyrer Res. 46, e96–e96. doi: 10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093 / bioinformatik / bty948
CrossRef Fuld tekst / Google Scholar
Kearse, M. G., J. E. (2017). Ikke-AUG oversættelse: en ny start for proteinsyntese i eukaryoter. Gener Dev. 31, 1717–1731. doi: 10.1101 / gad.305250.117
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Kong, L., Jang, Y., Ye, S.-K., Liu, S.-K., S.-K., V., L., et al. (2007). CPC: vurder proteinkodningspotentialet for udskrifter ved hjælp af sekvensfunktioner og supportvektormaskine. Nukleinsyrer Res. 35, 345–349. doi: 10.1093 / nar / gkm391
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Langmead, B., Salberg, S. L. (2012). Hurtig gapped-Læs justering med Butterfly 2. Nat. Metoder 9, 357-359. doi: 10.1038 / nmeth.1923
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
Meng, Chen, K., Yang, P., Chen, M. (2017). CircPro: et integreret værktøj til identifikation af circrna ‘ er med proteinkodningspotentiale. Bioinformatik 33, 3314-3316. doi: 10.1093 / bioinformatik / btks446
PubMed abstrakt / CrossRef fuldtekst / Google Scholar
Michel, A. M., Baranov, P. V. (2013). Ribosomprofilering: en Hi-Def-skærm til proteinsyntese i genom-bred skala: ribosomprofilering. Viley Interdiscip. Rev. RNA 4, 473-490. doi: 10.1002 / vrna.1172
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Jens, M., R., Stottmeister, C., Ruhe, L., Et Al. (2017). Oversættelse af CircRNAs. Mol. Celle 66, 9-21.e7. doi: 10.1016 / j. molcel.2017.02.021
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
Ponty, Y., Termier, M., Denise, A. (2006). GenRGenS: programmel til generering af tilfældige genomiske sekvenser og strukturer. Bioinformatik 22, 1534-1535. doi: 10.1093 / bioinformatik / btl113
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Rho, M., Tang, H., Ye, Y. (2010). FragGeneScan: forudsigelse af gener i korte og fejlbehæftede læsninger. Nukleinsyrer Res. 38, e191-e191. doi: 10.1093 / nar / 747
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
det er en af de bedste måder at gøre dette på. (2013). Peptidomisk opdagelse af korte åbne læserammekodede peptider i humane celler. Nat. Chem. Biol. 9, 59–64. doi: 10.1038 / nchembio.1120
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Spealman, P., Naik, A. V., May, G. E., Kuersten, S., Freeberg, L., Murphy, R. F., et al. (2018). Konserverede ikke-AUG uORFs afsløret ved en ny regressionsanalyse af ribosomprofileringsdata. Genom Res. 28, 214-222. doi: 10.1101 / gr.221507.117
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
Vicens, K., Vesthof, E. (2014). Biogenese af cirkulære RNA ‘ er. Celle 159, 13-14. doi: 10.1016 / j.celle.2014.09.005
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
det er en af de bedste måder at gøre dette på. (2017). N-terminal proteomics assisterede profilering af det uudforskede oversættelsesinitieringslandskab i Arabidopsis thaliana. Mol. Celle. Proteomics 16, 1064-1080. doi: 10.1074 / mcp.M116. 066662
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
Cai, T., Luo, J., VII, L. (2006). KOBAS server: en internetbaseret platform til automatisk annotation og vejidentifikation. Nukleinsyrer Res. 34, 720–724. doi: 10.1093 / nar / gkl167
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Yang, L., Fu, J., Jhou, Y. (2018). Cirkulære RNA ‘ er og deres nye roller i immunregulering. Front. Immunol. 9, 2977. doi: 10.3389 / fimmu.2018.02977
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar
Yang, Y., Fan, H., Mao, M., Song, H., P., Hang, Y., et al. (2017). Omfattende oversættelse af cirkulære RNA ‘ er drevet af N6-methyladenosin. Celle Res. 27, 626-641. doi: 10.1038 / cr.2017.31
PubMed Abstrakt / CrossRef Fuld Tekst / Google Scholar
Yu, G., Vang, L.-G., Han, Y., Han, K.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar