Úvod
svalové dystrofie končetin a pletenců (LGMDs) zahrnují heterogenní skupinu poruch charakterizovaných progresivním plýtváním a slabostí svalů proximálních končetin (1, 2). LGMDs vykazují inter a intrafamiliální variabilitu, od velmi mírných forem až po těžké, časné nástupy, rychle progresivní fenotypy (2). LGMDs lze klasifikovat jako autozomálně dominantní (LGMD1) nebo recesivní (LGMD2). První skupina má obvykle dospělý věk nástupu a není příliš častá (<10% všech Lgmd), zatímco druhá skupina je častější (1:15000) (1). Mezi recesivními formami je LGMD2A (nebo kalpainopatie) nejčastější LGMD na světě a postihuje přibližně 30% všech případů LGMDs (3). Mezi klinické příznaky onemocnění patří chůze po špičkách, chůze po kolenou, potíže s běháním a stoupáním po schodech, skapulární kyvadlo. Běžně se pozorují kontraktury kloubů, zkrácení Achillovy šlachy, skolióza, zatímco svaly obličeje a krku nejsou ovlivněny (4). Asymptomatická Hyperkémie (5-80násobek normálních hladin CPK) u mladých pacientů je považována za Předklinické stadium onemocnění a může přetrvávat několik let (1, 5). Věk nástupu svalové slabosti se obvykle vyskytuje ve věku 15 let, i když se může objevit v dřívějším (<12 let) nebo pozdějším (>30 let) věku. Progrese onemocnění může vést ke ztrátě ambulace, respirační nedostatečnosti a snížené vitální kapacitě plic v pokročilých stádiích (6). Srdeční postižení (poruchy srdečního rytmu ,poruchy srdečního vedení, dysfunkce ejekce levé komory) je hlášeno pouze příležitostně (6, 7).
diagnóza kalpainopatie je potvrzena detekcí patogenních mutací v CAPN3 (15q15.1) (4), kódujících různé alternativně sestříhané transkripty. Přepis v plné délce je však primárně exprimován ve svalové tkáni (8). Kódovaný protein (CAPN3) je členem rodiny cysteinových proteáz závislých na Lysozomálním Ca++. Ve svalech se CAPN3 účastní „remodelace sarkomerů“, která je nezbytná pro adaptaci a růst svalů v reakci na funkční a metabolické požadavky. K dnešnímu dni bylo popsáno více než 490 patogenních mutací v celém CAPN3, z nichž většina jsou jednonukleotidové změny (4). Mutace v CAPN3 byly spojeny s mitochondriálními abnormalitami, selháním růstu, zvýšeným oxidačním stresem a sarkomerovou dezorganizací, které celkově přispívají k tomu, že sval není schopen držet zátěž, což způsobuje degeneraci myofiber a ztrátu svalů (8). Diagnóza kalpainopatie může být náročná kvůli genetické heterogenitě a nespecifičnosti klinického a instrumentálního vzoru. Distribuce svalové slabosti / atrofie, hypertrofie/pseudo-hypertrofie a kontraktury šlach jsou velmi často sdíleny s jinými Lgmd nebo neuromuskulárními poruchami. Vzorek svalové biopsie u kalpainopatie je obecně také nespecifický, od mírných svalových abnormalit až po závažné dystrofické změny. Navíc imunohistochemické / biochemické markery obvykle nejsou spolehlivé: signál calpain může být normální i v přítomnosti nefunkčního proteinu a naopak může být redukován i u jiných svalových dystrofií odlišných od kalpainopatie.
proto se doporučuje provést diferenciální diagnostiku, aby byly poskytnuty přesné a spolehlivé výsledky. Za tímto účelem byly vyvinuty přístupy molekulárně genetického testování k potvrzení diagnózy kalpainopatie, včetně multigenního panelu, rozsáhlého genomu (sekvenování exomu/genomu) a analýzy jednoho genu (přímé sekvenování) (9). Implementace sekvenování nové generace (NGS) byla užitečná pro generování informativních dat, která mají být použita pro diagnostické, prediktivní nebo terapeutické účely (10-12). Genové panely NGS jsou založeny na analýze souboru genů spojených se specifickým onemocněním nebo skupinou souvisejících poruch, které jsou charakterizovány genetickou a fenotypovou heterogenitou. Panely NGS mohou být také užitečné pro detekci smíšených nebo komplexních fenotypů, které jsou výsledkem dědičnosti více než jednoho genetického defektu od rodičů (13). Obecně platí, že pacienti s komplexními fenotypy, kteří jsou negativní na známé mutace v panelech diagnostických genů navržených na zakázku a vyžadující širokou diferenciální diagnostiku, jsou způsobilí pro sekvenování celého exomu nebo genomu. Sekvenování celého exomu / genomu jsou dražší přístupy, pokud jde o správu dat, interpretaci a analytické náklady, ale zvyšují pravděpodobnost poskytnutí molekulární diagnostiky podezření na genetickou poruchu (13). Pokud jde o LGMDs, Vysoce se doporučují speciální panely NGS, aby byla zajištěna vysoká diagnostická rychlost, optimální pokrytí, citlivost a specificita klinického testování (9). Panely NGS proto představují jeden z nejlepších systémů pro usnadnění diferenciální diagnostiky, identifikaci nových kauzativních mutací a objasnění korelace genotyp-fenotyp (14, 15).
v tomto rukopisu je uveden případ pacienta postiženého LGMD2A, u kterého aplikace panelu NGS umožnila nejen potvrdit diagnózu kalpainopatie (mutace v CAPN3), ale také identifikovat další novou mutaci v genu LMNA spojenou s dilatační kardiomyopatií. Vzhledem k těmto výsledkům byla analýza rozšířena na rodinné příslušníky probandu, aby poskytla komplexnější interpretaci analytických dat ve vztahu k patologickým fenotypům.
prezentace případu
klinická charakterizace Probandu a příbuzných
nástup onemocnění v probandu byl uveden ve věku 10 let, kdy hlásila přítomnost hypertrofie lýtka s chůzí po špičkách, obtížemi při běhu,lezení po schodech a vstávání. Symptomy vykazovaly pomalé, ale progresivní zhoršení s následným postižením proximální horní končetiny. Ve věku 13 let byly náhodně objeveny vysoké hladiny CPK (~8 000 UI/L), které nikdy nebyly spojeny s myoglobinurií. Postupně pacient podstoupil několik neuromuskulárních vyšetření v různých specializovaných centrech. Byly provedeny dvě svalové biopsie, ukazující klasický dystrofický obraz (hypertrofická / atrofická vlákna, vnitřní jádra, nekrotická vlákna, zvýšení pojivové tkáně) s nespecifickými vlastnostmi. Podle očekávání byly imunochemické a imunoblotové studie neprůkazné, což naznačuje abnormální / redukovaný α, β, γ sarkoglykan, β-dystroglykan a dystrofinový signál pouze v nekrotických vláknech (opakovaná imunochemie vedla k normálu, také pro laminin); imunobloty pro dystrofin a kalpain odhalily normální signály. Je však známo, že imunoblot calpain-3 není zcela citlivý na diagnózu LGMD2A, protože 20-30% případů vykazuje normální množství proteinu (1, 16-18). Vzhledem ke klinické prezentaci (hyperckmie, hypertrofie lýtka/pseudo-hypertrofie) byly nejprve vyloučeny dystrofinopatie. Kromě toho analýza genů DMD (Xp21), FKRP (19q13.32) a DYSF (2p13.2) neodhalila patogenní mutace. Pacient přišel k našemu pozorování ve věku 30 let, představující axiální a pásové postižení jak v horní, tak v dolní končetině, s významnou chůzí a okřídlenými lopatkami. V dolních končetinách byla slabost prominentní u čtyřhlavého svalu a glutei svalů, které byly významně ipo-atrofické, spolu s důkazem „zjevně hypertrofické“ (pseudo-hypertrofie) lýtkových svalů (Obrázek 1). Křeče, myalgie a vlnění nebyly pozorovány. Kompletní pneumologické (spirometrie a polysomnografie) a kardiologické (echografie, 24-h monitorování EKG Holter, srdeční MRI, kompletní kardiologická fyzikální kontrola s cílenou anamnézou a kardiovaskulární reflexní analýzou) vyšetření neodhalily žádnou významnou abnormalitu. Vyloučili jsme také nedostatek kyselé maltázy (normální testy enzymové aktivity v lymfocytech / leukocytech). Magnetické rezonanční zobrazování svalů (MRI) prokázalo výrazné postižení skapulárního pletence, paravertebrálních svalů, zadních kompartmentových svalů stehna (s relativním šetřením svalů sartorius a gracilis) a nohy (zejména gastrocnemius medialis a soleus) (Obrázek 2). Tento vzorec zapojení již byl uveden v literatuře týkající se obrazu MRI u kalpainopatie (19, 20). Studie MRI navíc ukázala, že lýtkové svaly byly účinně atrofické a charakterizované infiltrací tuku, což způsobilo svalové „zvětšení“.“Klinická prezentace, věk nástupu, rychlost progrese, distribuce svalové slabosti a nálezy MRI v probandu byly plně v souladu s diagnózou LGMD2A podrobně popsanou v literatuře (4, 21, 22).
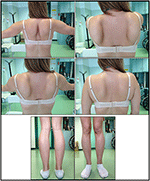
Obrázek 1. Proband klinické příznaky: okřídlené lopatky a kombinace atrofie stehna a pseudo-hypertrofie lýtka.
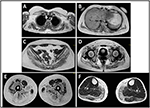
Obrázek 2. Proband svalové MRI: postižení pletence horní končetiny a paravertebrálních svalů (A,B), postižení pletence dolní končetiny a zadního kompartmentu stehna (C-E, většinou glutei) s relativním šetřením Sartorius a gracilis a postižení zadního kompartmentu nohy (F, většinou gastrocnemius medialis/soleus).
hodnocení rodinných příslušníků probandu neodhalilo žádné neuromuskulární onemocnění v anamnéze a žádné důkazy o příbuznosti mezi rodiči. Matka probandu však ve věku 55 let představila 2 synkopální epizody, u nichž byla diagnostikována idiopatická bradykardie(až 40 bpm v noci). Rozsáhlé kardiologické vyšetření na matce probandu diagnostikovalo symptomatický atrioventrikulární (AV) blok po výskytu několika synkopálních epizod / lipotymie. Analýza ukázala přítomnost základního AV bloku prvního stupně s několika obdobími těžké bradykardie, většinou noční a často symptomatické, spojené s některými fázemi AV bloku druhého stupně, typu 1 I 2, a některé fáze noční úplné av disociace. Echokardiografie a ergometrický test neodhalily významné změny, s výjimkou patentového foramen ovale. Matka nevykazovala žádný jiný klinický příznak, predispoziční stav ani rizikový faktor. Vzhledem k jejímu klinickému obrazu byla matce probandu implantována PMK. Kromě toho rodinná historie odhalila, že babička a pradědeček probandu byli také implantováni PMK ve věku 55 a 30 let. Navíc bratr babičky probanda zemřel v 51 letech na těžkou dilatační kardiomyopatii. Kardiologická vyšetření byla provedena také u Otce a strýce z matčiny strany, kteří byli zcela nedotčeni.
tato studie byla schválena etickou komisí nadace Santa Lucia a byla provedena podle Helsinské Deklarace. Všichni účastníci poskytli podepsaný informovaný souhlas s genetickou analýzou a v tomto ohledu také poskytli souhlas se zveřejněním této kazuistiky.
laboratorní vyšetření a diagnostické testy
genomová DNA byla extrahována z periferní krve (400 µL) pomocí Magpurix Blood DNA Extraction Kit a Magpurix Automatic Extraction System (Resnova) podle pokynů výrobce. Vzorky byly sekvenovány pomocí iontového PGM systému a Ion Ampliseq přizpůsobeného panelu s vysokou specificitou (Thermo Fisher Scientific). Velikost panelu byla 129.13 Kb, která se očekává, že screening ~99,72% z celkového panelu s minimálním pokrytím 20X. panel zahrnoval 18 genů, které byly vybrány pomocí vědecké literatury, GeneReviews (www.ncbi.nlm.nih.gov/books/NBK1116/) a četnost patogenních variant v běžné populaci. Podrobný popis panelu NGS byl shrnut v tabulce 1.
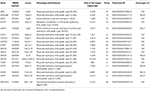
Tabulka 1. Přizpůsobený panel NGS používaný pro diagnostiku LGMD.
konstrukce knihoven byla provedena pomocí Ion AmpliSeq™ Library Kits 2.0. Pro multiplexní PCR reakce bylo použito přibližně 10 ng/µl výchozí DNA. Postupně byly provedeny dva kroky čištění (pomocí AMPure XP, Beckman Coulter) k odstranění nežádoucích kontaminantů a byla provedena konečná PCR. Template amplifikace a obohacování kroky byly provedeny Ion PGM Hi-Q OT2 kit-400, Ion OneTouch 2 Systém a Ion OneTouch ES (Thermo Fisher Scientific). Vzorky byly zpracovány Ion PGM Hi-Q Sequencing Kit (400 bp, Thermo Fisher Scientific) a běží na Ion 316 čip v2 (850 toky potřebné) a Ion PGM sekvencer (Thermo Fisher Scientific). Výsledky byly analyzovány pomocí Ion Reporter 4.6 (Thermo Fisher Scientific) a Integrated Genome Viewer (IGV). Interpretace genetických variant byla provedena pomocí Human Gene Mutation Database (HGMD), Leiden Open Variation Database (LOVD), ClinVar a ExAC. Funkční účinek detekovaných variant byl hodnocen pomocí bioinformatických prediktivních nástrojů, včetně mutace Taster, Varsome, SIFT, Polyfenum 2, SMART, Human Splicing Finder (HSF). Přímé sekvenování (BigDye Terminator v3.1, BigDyeX Terminator a ABI3130, Applied Biosystems) byl proveden pro potvrzení genetických variant a pro sekvenční genomické kódovací oblasti s pokrytím <20X (LMNA, Chr1:156106052-156106076; DYSF, Chr2: 71753352-71753502, Chr2: 71776385-71776640; SGCB, Chr4: 52904225-52904560; SGCA, Chr17: 48243242-48243570).
výsledky
analýza NGS probandu odhalila 3 heterozygotní varianty (Doplňkový Obrázek 1). V CAPN3 byly lokalizovány dvě varianty, a to NM_000070.2 (CAPN3): c. 550delA (s. Thr184Argfs) a c. 1813G>C (s. Val605Leu) v exonech 4 a 16. První je delece jednoho nukleotidu a je nejčastější (75% případů) patogenní variantou v evropských zemích (1). Podle očekávání bioinformatická analýza klasifikovala c. 550delA (rs80338800) jako variantu ztráty funkce (s. Thr184Argfs), což způsobilo posun snímků otevřeného čtecího rámce. Prediktivní nástroje (Mutation Taster, HSF, Varsome, PolyPhen 2, SIFT) popsaly variantu jako škodlivou. Kromě toho SMART odhalil, že změněný proteinový produkt postrádá doménu calpain 3 a tři motivy „EF-hands“. První doména je zapojena do signálních drah Kalpainu, zatímco EF-ruce jsou nezbytné pro aktivaci proteinu závislého na Ca++(4).
pokud jde o c. 1813G> C, Jedná se o novou variantu missense (str.Val605Leu), u které se předpokládalo, že je škodlivá (mutací degustátor, HSF, Varsome, Polyfen2, SIFT) kvůli potenciální změně sestřihu. Tato varianta není anotována v literatuře ani mezi online databázemi a nebyla nalezena u 200 kontrolních subjektů. Analýza SMART tool bohužel nepřinesla významné výsledky, protože varianta je umístěna v netypické doméně. Segregační analýza CAPN3 ukázala, že matka i strýc matky byli heterozygotní pro c. 550delA, zatímco otec byl nositelem c. 1813G>C.
kromě toho analýza NGS na proband odhalila přítomnost nové varianty v LMNA, jmenovitě NM_170707( LMNA): c. 550C>T (s. Gln184*). Výše uvedená varianta byla předpovězena jako nulová varianta, dosud nebyla popsána v literatuře ani mezi online databázemi a nebyla nalezena u 200 kontrolních subjektů. Prediktivní nástroje (Mutation Taster, HSF, Varsome, Polyfenum 2, SIFT) popsaly patogenní účinek této varianty na proteinový produkt. Inteligentní nástroj uvedl, že změněný protein LMNA postrádá doménu vlákna, což je nezbytné pro udržení jeho struktury a funkce. Segregační analýza zdůraznila přítomnost c.550C>t varianta pouze u matky, což naznačuje možnou souvislost s její srdeční symptomatologií a její rodinnou anamnézou onemocnění.
podle kritérií stanovených normami a pokyny American College of Medical Genetics (ACMG) (23), c. 1813G> C (CAPN3)lze popsat jako pravděpodobnou patogenní variantu vzhledem k tomu, že se nachází v kritické doméně bez benigní variace (PM1); chybí v databázích Exac, GnomAD a 1000 genom Browser databases (PM2).; vzhledem k recesivnímu modelu dědičnosti byl detekován v trans s patogenní variantou (PM3); více řádků výpočetních důkazů podporuje škodlivý účinek na gen nebo genový produkt (PP3). Pokud jde o klinickou klasifikaci c. 550C>T (LMNA) ACMG, může být označen jako patogenní varianta, protože se jedná o nulovou variantu způsobující ztrátu funkce (p. Gln184*) v LMNA (PVS1); chybí v databázích Exac, GnomAD a 1000 genomových prohlížečů (PM2); je umístěn v kritické doméně bez benigní variace (PM1).; více řádků výpočetních důkazů podporuje škodlivý účinek na gen nebo genový produkt (PP3).
závěry
tato kazuistika prezentovala pacienta postiženého LGMD, u kterého bylo zjištěno, že je nositelem mutací nejen v CAPN3 (c. 550delA a c. 1813G>C), ale také v LMNA (c. 550C>T). Tyto výsledky vysvětlují neuromuskulární fenotyp probandu kvůli mutacím CAPN3 a zdůrazňují potenciální riziko kardiovaskulárních poruch v důsledku přítomnosti varianty v LMNA a její pozitivní rodinné anamnéze. Vzhledem k těmto výsledkům byli členové rodiny podrobeni segregační analýze. Zejména matka měla za následek, že je nositelem CAPN3_c.550delA a LMNA_c.550C>T. Matka může být považována za zdravého nosiče patogenní mutace CAPN3_c.550delA spojené s kalpainopatií. Přítomnost nové varianty v LMNA však může vysvětlit její kardiovaskulární patologii (bradykardie a synkopální epizody). Absence neuromuskulární symptomatologie u matky a zvláštní klinický obraz probandu vyloučila možnou souvislost LMNA_c.550C> T s neuromuskulárním fenotypem, zejména pokud jde o Emery-Dreifussovu svalovou dystrofii (EMD). Ve skutečnosti EMD specificky ovlivňuje vastus lateralis a biceps brachii svaly, které jsou relativně ušetřeny v LGM2A (24, 25).
mateřský strýc byl heterozygotní pouze pro CAPN3_c. 550delA a podle očekávání nevykazoval žádné neuromuskulární nebo kardiovaskulární problémy. Otec měl za následek, že nesl román CAPN3_c. 1813G>C a tím nebyl ovlivněn. Celkově segregační analýza potvrdila dědičnost tří mutací probandu od jejích příbuzných a zdůraznila znalost kardiomyopatie ,kterou nelze zanedbat (obrázek 3).
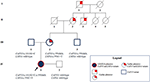
obrázek 3. Rodokmen ukazující pozitivní znalost srdečního fenotypu zděděného mateřskou linií a přenos variant CAPN3 a LMNA napříč členy rodiny.
celkově tato data vyvolávají některé důležité úvahy. Za prvé, panel NGS u tohoto pacienta byl rozhodující pro dosažení molekulární diagnózy kalpainopatie, detekce jedné známé mutace a druhé nové varianty předpovídané jako patogenní. Za druhé, panel NGS umožnil identifikaci nové varianty LMNA_c. 550C>T v probandu. Tento výsledek byl v souladu s pozitivní rodinnou anamnézou as údaji o segregaci, vysvětlením kardiovaskulárních onemocnění u matky a, co je důležitější, doporučením konkrétnějšího kardiologického sledování v probandu. Je důležité poznamenat, že kardiologické projevy se objevily u matky později ve věku (55 let), takže můžeme předpokládat, že proband může být stále v „asymptomatické kardiologické fázi.“Nelze však vyloučit variabilní fenotypovou expresi mutace LMNA v probandu ve srovnání s matkou. Všechny tyto údaje zdůrazňují význam integrovaného přístupu mezi lékaři a genetiky, pro správnou interpretaci výsledků, správné genetické poradenství a, případně, klinické řízení a sledování. Pokud jde o genetické a rodinné poradenství, analýza NGS byla také provedena na partnerovi probandu, aby se odhadlo reprodukční riziko pro pár. Partner byl negativní na všech 18 testovaných genů, což znamená, že má 1/650 zbytkové riziko, že bude zdravým nosičem kauzálních mutací LGMD, vzhledem k tomu, že test je 84% citlivý. S ohledem na genetický profil probandu a citlivost testu NGS je zbytkové riziko, že pár bude mít dítě postižené kalpainopatií, 1/1300. Na druhé straně jsou patogenní varianty LMNA přenášeny podle autozomálně dominantního vzoru, což znamená, že proband má 50% pravděpodobnost heterozygotních dětí. Klinický obraz potomstva však nelze jistě předvídat, protože lmna_c. 550C>T je nová varianta a jeho funkční dopad na fenotyp je stále neznámý.
závěrem tato kazuistika zdůrazňuje klinickou užitečnost panelů NGS poskytnout přesnou diagnózu LGMD2A a popsat komplexní fenotypy a komorbidity pocházející z dědičnosti různých mutací ve více genech. Aplikace NGS v klinické praxi by však měla být vždy kombinována s pre-a post-genetickým poradenstvím, aby bylo možné jasně vysvětlit výsledky, možné důsledky na fenotyp pacientů, riziko recidivy v rodině a vysvětlit možné neočekávané nálezy.
dostupnost dat
pro tuto studii nebyly generovány ani analyzovány žádné datové sady.
etické prohlášení
tato studie byla provedena v souladu s doporučeními Etické komise nadace Santa Lucia s písemným informovaným souhlasem všech subjektů. Všechny subjekty daly písemný informovaný souhlas v souladu s Helsinskou deklarací. Protokol byl schválen etickou komisí nadace Santa Lucia.
autorské příspěvky
RC, CSt, VC, GC, RG, GPa, SC, CP a JM přispěly k získávání dat, analýze a interpretaci dat. CSt, RC, GC, RG, GM a EG se podílely na přípravě rukopisu. SZ, GPr, CSa a SS se podílely na získávání klinických údajů. RC, CSt, VC, GC, RG, GPa, SC, CP, JM, SZ, GPr, GM, CSa, SS a EG udělily konečné schválení verze, která má být zveřejněna.
financování
tato práce je podporována ministerstvem zdravotnictví 5X 2016 2.
Prohlášení o střetu zájmů
autoři prohlašují, že výzkum byl proveden bez jakýchkoli obchodních nebo finančních vztahů, které by mohly být vykládány jako potenciální střet zájmů.
doplňkový materiál
doplňkový materiál k tomuto článku lze nalézt online na adrese: https://www.frontiersin.org/articles/10.3389/fneur.2019.00619/full#supplementary-material
1. Fanin M, Angelini C. Protein a genetická diagnostika svalové dystrofie končetin typu 2A: výnos a úskalí. Svalový Nerv. (2015) 52:163–73. doi: 10.1002 / mus.24682
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
2. Nigro V, Savarese m. genetický základ svalových dystrofií končetin a pletence: aktualizace 2014. Acta Myol. (2014) 33:1–12.
PubMed Abstrakt / Google Scholar
3. Richard I, Hogrel JY, Stockholm D, Payan CAM, Fougerousse F, Eymard B, et al. Přirozená historie LGMD2A pro vymezení výsledných opatření v klinických studiích. Ann Clintonová. (2016) 3:248–65. doi: 10.1002 / acn3.287
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
4. Angelini C, Fanin M. Calpainopathy. V: Adam MP, Ardinger HH, Pagon RA editors. GeneReviews®. Seattle, WA: University of Washington (2017) 1-30.
PubMed Abstrakt / Google Scholar
5. Kyriakides T, Angelini C, Schaefer J, Sacconi S, Siciliano G, Vilchez JJ, et al. Pokyny EFNS pro diagnostický přístup k pauci-nebo asymptomatické hyperkémii. Eur J Neurol. (2010) 17:767–73. doi: 10.1111 / j. 1468-1331. 2010. 03012.x
CrossRef Plný Text / Google Scholar
6. Mori-Jošimura M, Segawa K, Minami N, Oya Y, Komaki H, Nonaka I, et al. Kardiopulmonální dysfunkce u pacientů se svalovou dystrofií končetin a pletence 2A.svalový nerv. (2017) 55:465–9. doi: 10.1002 / mus.25369
CrossRef Plný Text / Google Scholar
7. Okere a, Reddy SS, Gupta S, Shinnar m. kardiomyopatie u pacienta se svalovou dystrofií končetin typu 2A. Circ srdeční selhání. (2013) 6: e12-13. doi: 10.1161 / CIRCHEARTFAILURE.112.971424
CrossRef Plný Text / Google Scholar
8. Kramerova I, Ermolova N, Eskin A, Hevener A, Quehenberger O, Armando AM, et al. Neschopnost up-regulovat transkripci genů nezbytných pro adaptaci svalů je základem svalové dystrofie 2A končetinového pletence (kalpainopatie). Hum Mol Genet. (2016) 25:2194–207. doi: 10.1093/hmg / ddw086
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
9. Thompson R, Straub V. svalové dystrofie končetin – mezinárodní spolupráce pro translační výzkum. Nat Rev Neurol. (2016) 12:294–309. doi: 10.1038 / nrneurol.2016.35
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
10. Strafella C, Caputo V, Galota MR, Zampatti S, Marella G, Mauriello S, et al. Aplikace přesné medicíny u neurodegenerativních onemocnění. Přední Neurol. (2018) 9:701. doi: 10.3389 / fneur.2018.00701
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
11. Cascella R, Strafella C, Caputo V, Errichiello V, Zampatti S, Milano F, et al. Směrem k aplikaci přesné medicíny při makulární degeneraci související s věkem. Prog Retin Eye Res. (2017) 63: 132-46. doi: 10.1016 / j. preteyeres.2017.11.004
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
12. Cascella R, Strafella C, Longo G, Manzo L, Ragazzo M, De Felici C, et al. Posouzení individuálního rizika pro AMD pomocí genetického poradenství, rodinné anamnézy a genetického testování. Oko. (2017) 32:446–50. doi: 10.1038 / oko.2017.192
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
13. Adams DR, Eng CM. Sekvenování nové generace k diagnostice podezření na genetické poruchy. N Engl J Med. (2019) 379:1353–62. doi: 10.1056 / NEJMc1814955
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
14. Angelini C. neuromuskulární onemocnění. Diagnostika a objev v svalové dystrofii končetin a pletence. Nat Rev Neurol. (2016) 12:6–8. doi: 10.1038 / nrneurol.2015.230
CrossRef Plný Text / Google Scholar
15. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, et al. Použití sekvenování celého exomu pro diagnostiku svalové dystrofie končetin a pletence: výsledky a získané poznatky. JAMA Neurol. (2015) 72:1424–32. doi: 10.1001 / jamaneurol.2015.2274
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
16. Milic A, Daniele N, Lochmüller H, Mora M, Comi GP, Moggio M, et al. Třetina biopsií LGMD2A má normální proteolytickou aktivitu calpain 3 stanovenou in vitro testem. Neuromusc Disord. (2007) 17:148–56. doi: 10.1016 / j. nmd.2006.11.001
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
17. Lanzillo R, Aurino S, Fanin M, Aguennoz M, Vitale F, Fiorillo C, et al. Časný nástup kalpainopatie s normální nefunkční hladinou kalpain 3. Dev Med Dětský Neurol. (2006) 48:304–6. doi: 10.1017 / S001216220600065X
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
18. Talim B, Ognibene A, Mattioli E, Richard I, Anderson LV, Merlini L. normální exprese kalpainu u geneticky potvrzené svalové dystrofie končetin a pletence typu 2A. neurologie. (2001) 56:692–3. doi: 10.1212 / wnl.56.5.692-a
CrossRef Plný Text / Google Scholar
19. Díaz-Manera J, Llauger J, Gallardo E, Illa i. svalová MRI u svalových dystrofií. Acta Myol. (2015) 34:95–108.
PubMed Abstrakt / Google Scholar
20. Mercuri E, Bushby K, Ricci E, Birchall DR, Pane M, Kinali M, et al. Nálezy MRI svalů u pacientů se svalovou dystrofií pletence končetin s deficitem kalpain 3 (LGMD2A) a časnými kontrakturami. Neuromusc Disord. (2005) 15:164–71. doi: 10.1016 / j. nmd.2004.10.008
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
21. Magri F, Nigro V, Angelini C, Mongini T, Mora M, Moroni I, et al. Italský registr svalové dystrofie pletence končetin: relativní frekvence, klinické příznaky a diferenciální diagnostika. Svalový Nerv. (2017) 55:55–68. doi: 10.1002 / mus.25192
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
22. Chae J, Minami N, Jin Y, Nakagawa M, Murayama K, Igarashi F, et al. Calpain 3 genové mutace: genetické a klinicko-patologické nálezy u svalové dystrofie končetin a pletence. Neuromusc Disord. (2001) 11:547–55. doi: 10.1016/S0960-8966(01)00197-3
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
23. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standardy a pokyny pro interpretaci sekvenčních variant: společné konsensuální doporučení Americké vysoké školy lékařské genetiky a genomiky a Asociace pro molekulární patologii. Genet Med. (2015) 17:405–24. doi: 10.1038 / gim.2015.30
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
24. Bonne G, Mercuri E, Muchir A, Urtizberea A, Bécane HM, Recan D, et al. Klinické a molekulárně genetické spektrum autozomálně dominantní Emery-Dreifussovy svalové dystrofie v důsledku mutací genu lamin A / C. Ann Neurolová. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170::podpora-ANA6>3.0.CO;2-J
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
25. Hong JS, Ki CS, Kim JW, Suh YL, Kim JS, Baek KK, et al. Srdeční dysrytmie, kardiomyopatie a svalová dystrofie u pacientů se svalovou dystrofií Emery-Dreifuss a svalovou dystrofií končetin a pletence typu 1B.J Korean Med Sci. (2005) 20:283–90. doi: 10.3346 / jkms.2005.20.2.283
CrossRef Full Text / Google Scholar