- Úvod
- metody
- filtrování ribozomálních profilovacích dat
- sestavování virtuálních genomů
- stanovení read-mapované oblasti Ribo-seq na křižovatce (RMRJ) circrna
- školení modelu a klasifikace RMRJs
- predikce přeložených peptidů pomocí RMRJs
- výsledky a diskuse
- přeložené cirkrna u lidí a a. thaliana
- funkční obohacení cirkrna člověka a a. thaliana s kódovacím potenciálem
- test přesnosti pro CircCode
- vliv hloubky sekvenování dat Ribo-seq
- porovnání CircCode s jinými nástroji
- závěry
- Dostupnost a požadavky
- Prohlášení o dostupnosti dat
- autorské příspěvky
- financování
- střet zájmů
- doplňkový materiál
Úvod
kruhové RNA (circrna) jsou zvláštním typem nekódující molekuly RNA, která se stala horkým výzkumným tématem v oblasti RNA a dostává velkou pozornost (Chen a Yang, 2015). Ve srovnání s tradičními lineárními RNA (obsahujícími 5′ a 3′ konce) mají molekuly circRNA obvykle uzavřenou kruhovou strukturu; což je činí stabilnějšími a méně náchylnými k degradaci (Vicens and Westhof, 2014). Ačkoli existence cirkrna je již nějakou dobu známa, tyto molekuly byly považovány za vedlejší produkt sestřihu RNA. S vývojem vysoce výkonných sekvenačních a bioinformatických technologií se však cirkrna staly široce uznávanými u zvířat a rostlin (Chen a Yang, 2015). Nedávné studie také ukázaly, že velké množství cirkrna může být přeloženo do malých peptidů v buňkách (Pamudurti et al., 2017) a mají klíčové role i přes jejich někdy nízkou úroveň výrazu (Hsu a Benfey, 2018; Yang et al. , 2018). Přestože se identifikuje rostoucí počet cirkrna, jejich funkce v rostlinách a zvířatech obecně zůstávají studovány. Kromě svých funkcí jako návnad miRNA mají cirkrna důležitý translační potenciál,ale nejsou k dispozici žádné nástroje pro specifické předpovídání translačních schopností těchto molekul (Jakobi a Dieterich, 2019).
existuje několik nástrojů pro predikci a identifikaci circrna, jako je CIRI (Gao et al., 2015), CIRCexplorer (Dong et al., 2019), CircPro (Meng et al., 2017) a circtools (Jakobi et al., 2018). Mezi nimi může CircPro odhalit přeložené circrna výpočtem skóre translačního potenciálu pro circrna na základě CPC (Kong et al ., 2007), což je nástroj pro identifikaci otevřeného čtecího rámce (ORF) v daném pořadí. Nicméně, protože některé circrna nepoužívají počáteční kodon během překladu (Ingolia et al., 2011; Slavoff a kol., 2013; Kearse a Wilusz, 2017; Spealman a kol., 2018), zaměstnávající CPC může odfiltrovat některé skutečně přeložené cirkrna. V této studii jsme použili BASiNET (Ito et al., 2018), což je RNA klasifikátor založený na metodách strojového učení (random forest a J48 model). Nejprve transformuje dané kódující RNA (pozitivní data) a nekódující RNA (negativní data) a reprezentuje je jako komplexní sítě; poté extrahuje topologická opatření těchto sítí a konstruuje vektorový prvek pro výcvik modelu, který se používá ke klasifikaci kódovací kapacity cirkrna. Při této metodě se zabrání chybnému filtrování přeložených cirkrna, které nejsou iniciovány AUG. Navíc technologie Ribo-seq, která je založena na vysoce výkonném sekvenování pro sledování RPFs (ribozomálně chráněné fragmenty) transkriptů (Guttman et al., 2013; Brar a Weissman, 2015), lze použít k určení umístění circrna, které jsou překládány (Michel a Baranov, 2013). Identifikovat kódovací schopnost circrna, vyvinuli jsme nástroj CircCode, který zahrnuje rámec Python 3 založený, a aplikovaný CircCode pro zkoumání překladového potenciálu circrna od lidí a Arabidopsis thaliana. Naše práce poskytuje bohatý zdroj pro další studium funkcí cirkrna s kódovací kapacitou.
metody
CircCode byl napsán v programovacím jazyce Python 3; používá Trimmomatic (Bolger et al., 2014), bowtie (Langmead a Salzberg, 2012) a STAR (Dobin et al., 2013) filtrovat syrové Ribo-seq čtení a mapovat tyto filtrované čtení do genomu. CircCode pak identifikuje Ribo-seq read mapované oblasti v circrna, které obsahují křižovatky. Poté jsou kandidátské mapované sekvence v circrna řazeny na základě klasifikátorů (model J48) do kódujících RNA a nekódujících RNA Basinetem. Nakonec jsou krátké peptidy produkované translací identifikovány jako potenciální kódující oblasti cirkrna. Celý proces CircCode se skládá z pěti kroků (Obrázek 1).
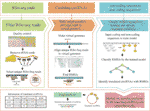
figura 1 pracovní postup CircCode. Horní vrstva představuje vstupní soubor potřebný pro každý krok CircCode. Střední vrstva je rozdělena na tři části a každá část představuje jinou fázi provozu. Zleva doprava, první část představuje filtrování dat Ribo-seq; kontrola kvality je prováděna Trimmomatic, a čtení rRNA jsou odstraněny motýlkem. Druhá část představuje kroky použité k vytvoření virtuálního genomu a zarovnání filtrovaných čtení s virtuálním genomem s hvězdou. Poslední část představuje identifikaci přeložených cirkrna pomocí strojového učení. Spodní vrstva představuje poslední krok použitý k predikci peptidů přeložených z cirkrna a konečných výstupních výsledků, včetně informací o přeložených cirkrna a jejich překladových produktech.
filtrování ribozomálních profilovacích dat
nejprve jsou fragmenty a adaptéry nízké kvality ve čtení Ribo-Seq odstraněny Trimmomaticem s výchozími parametry, aby se dosáhlo čistého čtení Ribo-seq. Za druhé, tyto čisté Ribo-seq čtení jsou mapovány do knihovny rRNA k odstranění čtení odvozených z rRNA pomocí bowtie. Protože délky čtení Ribo-seq jsou relativně krátké (obecně méně než 50 bp), je možné, aby jedno čtení odpovídalo více regionům. V tomto případě je obtížné určit, které oblasti konkrétní čtení odpovídá. Aby se tomu zabránilo, čistá čtení Ribo-seq jsou mapována na genom sledovaného druhu,a čtení, která nejsou dokonale zarovnána s genomem, jsou považována za konečné jedinečné čtení Ribo-seq.
sestavování virtuálních genomů
Cirkrna se obvykle objevují jako molekuly ve tvaru prstence v eukaryotách a mohou být identifikovány na základě jejich zpětných spojů. Sekvence cirkrna v souboru fasta jsou však často v lineární formě. Teoreticky, výsledek naznačuje, že spojení je mezi 5′ terminálním nukleotidem a 3′ terminálním nukleotidem, ačkoli křižovatku a sekvenci v blízkosti křižovatky nelze vidět přímo, tak zarovnání Ribo-seq čte na circRNA sekvence, včetně křižovatek, přímým způsobem.
CircCode spojuje sekvenci každé circRNA v tandemu tak, že křižovatka pro každou je uprostřed nově vytvořené sekvence. Každou sériovou jednotku jsme také oddělili 100 N nukleotidy, abychom se vyhnuli záměně v kroku zarovnání sekvence (délka každého RPF je menší než 50 bp). Nakonec jsme získali virtuální genom sestávající pouze z kandidátských cirkrna v tandemu oddělených 100 Ns. Protože CircCode se zaměřuje pouze na zarovnání mezi Ribo-seq čtením a circrna sekvencemi, můžeme zkoumat kódovací potenciál circrna mapováním Ribo-seq čtení na tento virtuální genom, což může ušetřit velké množství výpočetního času (virtuální genom je mnohem menší než celý genom) a zvýšit přesnost (tím, že se vyhneme interferenci mezi porovnáním circrna proti proudu a po proudu).
stanovení read-mapované oblasti Ribo-seq na křižovatce (RMRJ) circrna
konečné jedinečné čtení Ribo-seq jsou mapovány na dříve vytvořený virtuální genom pomocí STAR. Protože každá tandemová cirkrna jednotka byla před vytvořením virtuálního genomu oddělena 100 N bázemi, byla největší délka intronu nastavena tak, aby nepřekročila 10 bází s parametrem “ – alignIntronMax 10.“Tento parametr eliminuje jakoukoli interakci mezi různými cirkrna v zarovnání sekvence. Ve druhém kroku produkce virtuálního genomu ukládá CircCode informace o pozičním spojení pro každou circRNA ve virtuálním genomu. V případě, že Ribo-seq read mapované oblasti ve virtuálním genomu zahrnuje křižovatku circRNA, a počet mapovaných Ribo-seq čte na křižovatce (NMJ) je větší než 3, Ribo-seq read mapované oblasti na křižovatce circrna lze považovat za RMRJ, který odhaluje zhruba přeložený segment circrna v blízkosti místa křižovatky.
školení modelu a klasifikace RMRJs
ačkoli RMRJs může představovat silný důkaz překladu, v této metodě stále existují určité nedostatky. Vzhledem k tomu, že délka čtení ribozomální mapy je krátká, lze čtení porovnat se špatnou polohou. Proto není přesvědčivé jednoduše považovat region, na který se vztahuje Ribo-seq, za přeložený region. Za tímto účelem se metoda strojového učení používá k identifikaci kódovací schopnosti RMRJ. Nejprve CircCode extrahuje kódující RNA (pozitivní data) a nekódující RNA (negativní data) z zájmového druhu a používá je pro modelový trénink pomocí rozdílu ve vektorech znaků mezi kódováním A nekódujícími RNA. CircCode pak používá vyškolený model ke klasifikaci RMRJs získaných v předchozím kroku BASiNET. Pokud je RMRJ circRNA rozpoznána jako kódující RNA, lze tuto circRNA identifikovat jako přeloženou circRNA.
predikce přeložených peptidů pomocí RMRJs
vzhledem k tomu, že exprese cirkrna v organismech je nízká, údaje Ribo-seq neukazují přesnou periodicitu 3-nt jasně v případě menšího počtu RPF. Proto, je obtížné určit přesné místo začátku překladu přeložené circRNA. Vzhledem k přítomnosti stop kodonu v některých RMRJs a vzhledem k tomu, že start kodon je obtížné určit, způsob nalezení ORF na základě start kodonu a stop kodonu není proveditelný.
určit skutečné Překladové oblasti těchto circrna a vygenerovat konečný překladový produkt, FragGeneScan (Rho et al., 2010), který může předpovídat oblasti kódující proteiny v fragmentovaných genech a genech s rámcovými posuny, se používá k určení přeložených peptidů produkovaných circrna.
aby se zabránilo těžkopádnému běžícímu procesu, mohou být všechny modely vyvolány shellovým skriptem; uživatel může jednoduše vyplnit daný konfigurační soubor a vložit jej do skriptu a poté bude spuštěn celý proces předpovídání přeložených circrna. Kromě toho může být CircCode spuštěn samostatně, krok za krokem, takže uživatel může upravit parametry uprostřed postupu a zobrazit výsledky každého kroku podle potřeby.
výsledky a diskuse
po testování na více počítačích bylo zjištěno, že CircCode úspěšně běží s nainstalovanými požadovanými závislostmi. K testování výkonu CircCode jsme použili data pro člověka a a. thaliana k předpovědi cirkrna s překladovým potenciálem. Výsledky byly porovnány s circrna, které byly experimentálně ověřeny jako potvrzení. Poté jsme dále testovali hodnotu false discovery rate (FDR) CircCode. Použili jsme GenRGenS (Ponty et al., 2006) vygenerovat soubor dat pro testování na základě známých přeložených circrna a potvrdil, že hodnota FDR byla v přijatelném rozmezí a na nízké úrovni. Nakonec jsme vyhodnotili vliv různých hloubek sekvenování dat Ribo-seq na předpovědi CircCode a porovnali CircCode s jiným softwarem.
přeložené cirkrna u lidí a a. thaliana
Chcete-li použít nástroj CircCode na skutečná data, nejprve jsme stáhli soubory včetně lidského referenčního genomu GRCh38, anotace genomu a lidské rRNA z Ensembl. Pro A. thaliana byly všechny referenční genomy (TAIR10), soubory anotace genomu a odpovídající sekvence rRNA staženy z rostlin Ensembl. Údaje Ribo – seq pro člověka a a. thaliana byly staženy z RPFdb (přístupová čísla: GSE96643, GSE81295, GSE88794) (Hsu et al., 2016; Willems et al., 2017), a všechny kandidátské okruhy z human A a. thaliana byly staženy z CIRCPedia v2 (Dong et al ., 2018) a PlantcircBase (Chu et al., 2017). Nakonec jsme identifikovali 3 610 přeložených cirkrna od člověka a 1 569 přeložených cirkrna od A. thaliana pomocí CircCode (doplňkové údaje 1).
funkční obohacení cirkrna člověka a a. thaliana s kódovacím potenciálem
pomocí výsledků CircCode pro člověka a a. thaliana, online nástroj KOBAS 3.0 (Wu et al ., 2006) byl použit k anotaci těchto přeložených cirkrna na základě jejich mateřských genů. Dále jsme provedli funkční analýzu GO (genová Ontologie) a analýzu obohacení KEGG (Kjótská encyklopedie genů a genomů) pro tyto přeložené circrna pomocí balíčku r clusterProfiler (Yu et al., 2012).
výsledky KEGG ukázaly, že lidské cirkrna byly obohaceny o zpracování bílkovin v endoplazmatické dráze retikula, dráze metabolismu uhlíku a transportní dráze RNA. GO analýza ukázala účast lidských přeložených cirkrna na regulaci vazby molekul, atpázové aktivity a dalších biologických procesů souvisejících s sestřihem RNA. Kromě toho jsou přeložené cirkrny a. thaliana obohaceny o cesty související s odolností proti stresu, což naznačuje, že v tomto procesu hrají zásadní roli (doplňkové údaje 2).
test přesnosti pro CircCode
ke zkoumání přesnosti CircCode byly použity testovací sekvence generované Genrgensem, který používá skrytý Markovův model k produkci sekvencí, které mají stejné sekvenční charakteristiky (jako jsou frekvence různých nukleotidů, různých kodonů a různých nukleotidů na začátku sekvence).
pro tuto studii jsme použili dříve publikované lidské přeložené circrna (Yang et al., 2017) jako vstup pro GenRGenS a vygeneroval 10 000 sekvencí pro testování CircCode. Test jsme opakovali 10krát a v průměru bylo pokaždé předpovězeno 27 přeložených cirkrna. Hodnota FDR byla vypočtena na 0,0027, což je mnohem méně než 0,05, což naznačuje, že předpokládané výsledky jsou věrohodné.
kromě toho jsme porovnali přeložené cirkrna od lidí identifikované pomocí CircCode s ověřenými daty cirkrna spojenými s polysomem (Yang et al ., 2017). Mezi nimi bylo 60% cirkrna identifikováno pomocí CircCode (doplňkové údaje 3).
vliv hloubky sekvenování dat Ribo-seq
abychom zkoumali dopad hloubky sekvenování dat Ribo-seq na výsledky identifikace CircCode, nejprve jsme testovali vliv hloubky sekvenování na počet přeložených circrna (obrázek 2A). Když byla hloubka sekvenování nízká, předpokládaný počet přeložených cirkrna byl nízký a počet přeložených cirkrna se zvyšoval se zvyšující se hloubkou sekvenování. Počet přeložených cirkrna se stal stabilním, když hloubka sekvenování dosáhla nejméně 10× pokrytí lineárním přepisem.

Obrázek 2 (A) vliv hloubky sekvenování dat Ribo-seq na předpokládaný počet přeložených cirkrna. (B) vliv Junction read number (JRN) na citlivost CircCode v různých hloubkách sekvenování.
za druhé byl také hodnocen vliv NMJ na citlivost v různých hloubkách sekvenování (obrázek 2B). Výsledky ukázaly, že NMJ měl menší dopad na citlivost, protože se zvyšovala hloubka sekvenování. CircCode měl také vyšší citlivost při použití dat Ribo-seq s vyšší hloubkou sekvenování.
porovnání CircCode s jinými nástroji
pro porovnání CircCode s jinými nástroji, jako je CircPro, byla stejná sada dat Ribo-seq (SRR3495999) od A. thaliana použita k identifikaci přeložených circrna pomocí šesti procesorů se 16 gigabajty paměti RAM. CircPro identifikoval 44 přeložených cirkrna za 13 minut, zatímco CircCode identifikoval 76 přeložených cirkrna za 20 minut. CircCode je tedy citlivější než CircPro na stejné úrovni hardwaru počítače, ale trvá to více času. CircPro je stručné a méně časově náročné než CircCode, ale CircCode může identifikovat více circrna s kódovací schopností než CircPro.
závěry
Cirkrna hrají důležitou roli v biologii a je zásadní přesně identifikovat cirkrna s kódovací schopností pro následný výzkum. Na základě Pythonu 3 jsme vyvinuli CircCode, snadno použitelný nástroj příkazového řádku, který má vysokou citlivost pro identifikaci přeložených circrna z čtení Ribo-Seq s vysokou přesností. CircCode vykazuje dobrý výkon jak u rostlin, tak u zvířat. Budoucí práce přidá následnou analýzu znaků do CircCode vizualizací každého kroku procesu a optimalizací přesnosti predikce.
Dostupnost a požadavky
CircCode je k dispozici na https://github.com/PSSUN/CircCode; operační systém(y): Linux, programovací jazyky: Python 3 A R; další požadavky: bedtools (verze 2.20.0 nebo novější), bowtie, STAR, Python 3 balíčky (Biopython, pandy, rpy2), R-balíčky (BASiNET, Biostrings). Instalační balíčky pro veškerý požadovaný software jsou k dispozici na domovské stránce CircCode. Uživatelé je nemusí stahovat jednotlivě. Domovská stránka CircCode také poskytuje podrobné uživatelské příručky pro referenci. Nástroj je volně k dispozici. Neexistují žádná omezení pro použití nonacademics.
Prohlášení o dostupnosti dat
všechna relevantní data jsou v rukopisu a jeho podpůrných informačních souborech.
autorské příspěvky
konceptualizace: PS, GL. Data Curation: PS, GL. Formální analýza: PS, GL. Psaní-původní návrh: PS, GL. Psaní-recenze a editace: PS, GL.
financování
tato práce byla podpořena granty z National Natural Science Foundation of China (grant č. 31770333, 31370329, a 11631012), Program pro nové století vynikající talenty na univerzitě (NCET-12-0896), a základní výzkumné fondy pro centrální univerzity (no. GK201403004). Finanční agentury neměly žádnou roli ve studii, její design, sběr a analýza dat, rozhodnutí o zveřejnění, nebo příprava rukopisu. Investoři neměli žádnou roli při návrhu studie, sběr a analýza dat, rozhodnutí o zveřejnění, nebo příprava rukopisu.
střet zájmů
autoři prohlašují, že výzkum byl proveden bez jakýchkoli obchodních nebo finančních vztahů, které by mohly být vykládány jako potenciální střet zájmů.
doplňkový materiál
doplňkový materiál k tomuto článku lze nalézt online na adrese: https://www.frontiersin.org/articles/10.3389/fgene.2019.00981/full#supplementary-material
doplňující údaje 1 / sekvence predikované přeložené circRNA a krátkého peptidu.
doplňující údaje 2 / GO enrichment and Kegg enrichment results for humans and Arabidopsis thaliana.
doplňující údaje 3 / srovnání předpokládaných přeložených cirkrna s validovanými přeloženými cirkrna.
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: flexibilní zastřihovač pro sekvenční data illumina. Bioinformatika 30, 2114-2120. doi: 10.1093 / bioinformatika / btu170
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Brar, g. a., Weissman, J. S. (2015). Profilování ribozomu odhaluje co, kdy, kde a jak syntézy proteinů. Adresa. Reverend Mol. Cell Biol. 16, 651–664. doi: 10.1038 / nrm4069
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Chen, L. – L., Yang, L. (2015). Regulace biogeneze circRNA. RNA Biol. 12, 381–388. doi: 10.1080/15476286.2015.1020271
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
chu, Q., Zhang, X., Zhu, X., Liu, C., Mao, L., Ye, C., et al. (2017). PlantcircBase: databáze pro kruhové RNA rostlin. Molo. Závod 10, 1126-1128. doi: 10.1016 / j. molp.2017.03.003
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Dobin, a., Davis, C. a., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). Hvězda: ultrarychlý univerzální RNA-seq aligner. Bioinformatika 29, 15-21. doi: 10.1093 / bioinformatika / bts635
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Dong, r., Ma, X. – K., Chen, L. – L., Yang, L. (2019). „Genome-wide anotace circrna a jejich alternativní back-sestřih / sestřih s CIRCexplorer potrubí,“ v Epitranscriptomics. EDA. Wajapeyee, N., Gupta, R. (New York, NY: Springer New York), 137-149. doi: 10.1007/978-1-4939-8808-2_10
CrossRef Plný Text / Google Scholar
Dong, r., Ma, X. – k., Li, G. – W., Yang, L. (2018). CIRCpedia v2: aktualizovaná databáze pro komplexní anotaci kruhové RNA a srovnání výrazů. Genomika Proteomika Bioinf. 16, 226–233. doi: 10.1016 / j. gpb.2018.08.001
CrossRef Plný Text / Google Scholar
Gao, Y., Wang, J., Zhao, F. (2015). CIRI: efektivní a nezaujatý algoritmus pro de novo identifikaci kruhové RNA. Genom Biol. 16, 4. doi: 10.1186 / s13059-014-0571-3
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Guttman, m., Russell, P., Ingolia, N. T., Weissman, J. S., Lander, E. S. (2013). Profilování ribozomů poskytuje důkaz, že velké nekódující RNA nekódují proteiny. Cela 154, 240-251. doi: 10.1016 / j. buňka.2013.06.009
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Hsu, P. Y., Benfey, P. N. (2018). Malý, ale mocný: funkční peptidy kódované malými Orfy v rostlinách. Proteomika 18, 1700038. doi: 10.1002 / pmic.201700038
CrossRef Plný Text / Google Scholar
Hsu, P. Y., Calviello, L., Wu, H.-Y. L., Li, F.-W., Rothfels, C. J., Ohler, U., et al. (2016). Profilování ribozomů se Super rozlišením odhaluje neoznámené translační události v Arabidopsis. Proc. Natle. Acad. Věda. 113, E7126-E7135. doi: 10.1073 / pnas.1614788113
CrossRef Plný Text / Google Scholar
Ingolia, N. T., Lareau, L. F., Weissman, J. S. (2011). Ribozomové profilování myších embryonálních kmenových buněk odhaluje složitost a dynamiku savčích proteomů. Cela 147, 789-802. doi: 10.1016 / j. buňka.2011.10.002
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
ito, E. a., Katahira, i., Vicente, F. F., da, R., Pereira, L. F. P., Lopes, F. M. (2018). BASiNET-biologická sekvenční síť: případová studie Kódování A nekódující identifikace RNA. Nukleové kyseliny rez.46, e96–e96. doi: 10.1093/nar/gky462
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Dieterich, C. (2019). Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA,10 (3), e1528. doi: 10.1002/wrna.1528
PubMed Abstract | CrossRef Full Text | Google Scholar
Jakobi, T., Uvarovskii, A., Dieterich, C. (2018). circtools—a one-stop software solution for circular RNA research. Bioinformatics 35 (13), 2326–2328. doi: 10.1093 / bioinformatika / bty948
CrossRef Plný Text / Google Scholar
Kearse, M. G., Wilusz, je (2017). Non-AUG translation: nový začátek syntézy proteinů u eukaryot. Geny Dev. 31, 1717–1731. doi: 10.1101 / gad.305250.117
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Kong, L., Zhang, y., Ye, z.-Q., Liu, x.-q., Zhao, s.-Q., Wei, L., et al. (2007). CPC: posoudit potenciál transkriptů kódujících proteiny pomocí sekvenčních funkcí a podpůrného vektorového stroje. Nukleové Kyseliny Rez.35, W345–W349. doi: 10.1093/nar / gkm391
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Langmead, B., Salzberg, S. L. (2012). Fast gapped-čtení zarovnání s Bowtie 2. Adresa. Metody 9, 357-359. doi: 10.1038 / nmeth.1923
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Meng, X., Chen, Q., Zhang, P., Chen, M. (2017). CircPro: integrovaný nástroj pro identifikaci circrna s potenciálem kódování proteinů. Bioinformatika 33, 3314-3316. doi: 10.1093 / bioinformatika / btx446
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Michel, a. M., Baranov, P.V. (2013). Profilování ribozomu: Hi-Def monitor pro syntézu proteinů v celém genomu: profilování ribozomu. Wiley Interdiscip. Rev. RNA 4, 473-490. doi: 10.1002 / wrna.1172
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Pamudurti, n. r., Bartok, o., Jens, m., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., et al. (2017). Překlad Cirkrna. Molo. Cela 66, 9-21.e7. doi: 10.1016 / j. molcel.2017.02.021
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Ponty, Y., Termier, M., Denise, A. (2006). GenRGenS: software pro generování náhodných genomových sekvencí a struktur. Bioinformatika 22, 1534-1535. doi: 10.1093 / bioinformatika / btl113
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Rho, m., Tang, h., Ye, y. (2010). FragGeneScan: předpovídání genů v krátkých a náchylných k chybám. Nukleové kyseliny rez.38, e191–e191. doi: 10.1093/nar / gkq747
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Slavoff, s. a., Mitchell, A. J., Schwaid, a. G., Cabili, m. n., Ma, J., Levin, J. Z., et al. (2013). Peptidomický objev krátkých peptidů kódovaných v otevřeném čtecím rámečku v lidských buňkách. Adresa. Cheme. Biol. 9, 59–64. doi: 10.1038/nchembio.1120
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Spealman, P., Naik, A. W., May, G. E., Kuersten, s., Freeberg, L., Murphy, R. F., et al. (2018). Konzervované non-AUG uORFs odhalené novou regresní analýzou profilovacích dat ribozomu. Genome Res.28, 214-222. doi: 10.1101 / gr.221507.117
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Vicens, Q., Westhof, E. (2014). Biogeneze kruhových RNA. Cela 159, 13-14. doi: 10.1016 / j. buňka.2014.09.005
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Willems, P., Ndah, e., Jonckheere, v., Stael, s., Sticker, a., Martens, L., et al. (2017). N-terminální proteomika napomohla profilování neprozkoumané krajiny iniciace překladu v Arabidopsis thaliana. Molo. Buňka. Proteomika 16, 1064-1080. doi: 10.1074 / mcp.M116. 066662
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Wu, J., Mao, X., Cai, T., Luo, J., Wei, L. (2006). KOBAS server: webová platforma pro automatickou anotaci a identifikaci cest. Nukleové Kyseliny Rez.34, W720–W724. doi: 10.1093/nar / gkl167
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
jang, L., Fu, J., Zhou, y. (2018). Kruhové RNA a jejich vznikající role v imunitní regulaci. Před. Immunol. 9, 2977. doi: 10.3389 / fimmu.2018.02977
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Yang, y., Fan, X., Mao, m., Song, X., Wu, P., Zhang, y. a kol. (2017). Rozsáhlá translace kruhových RNA poháněných N6-methyladenosinem. Cell Res.27, 626-641. doi: 10.1038 / cr.2017.31
PubMed Abstrakt / CrossRef Plný Text / Google Scholar
Yu, G., Wang, L.-G., Han, Y., He, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
CrossRef Full Text | Google Scholar